

CLINICAL LABORATORY DIAGNOSTICS OF ANEMIAS
In its broadest sense, anemia is a functional inability of the blood to supply the tissue with adequate O2 for proper metabolic function. Anemia is not a disease, but rather the expression of an underlying disorder or disease. Anemia is usually associated with decreased levels of hemoglobin and/or a decreased packed cell volume (hematocrit), and/or a decreased RBC count.
Classification: There are two main ways to classify the anemias:
a. Morphologicall (based on red cell size and hemoglobin content)
(1) Microcytic hypochromic
(MCV < 80 mcm 3, diameter of erythrocyte < 6,5 mcm)
(2) Macrocytic hyperchromic
(MCV - mean corpuscular volume >100 mcm3, diameter of erythrocyte > 8 mcm)
(3) Normocytic normochromic
(MCV = 81-99 mcm 3, diameter of erythrocyte = 7,2-7,5 mcm)
b. Pathophysiological (based on causative factors)
(1) Blood Loss
(2) Impaired RBC production
(3) Increased RBC destruction
Pathophysiological Classification of Anemias
I. Blood Loss Anemia
A. Acute posthemorrhagic anemia
B. Chronic posthemorrhagic anemia
II. Impaired RBC Production
A. Disturbance of the proliferation and differentiation of stem cells
1. Multipotential stem cells
a. Aplastic anemia
2. Unipotential stem cells
a. Pure red cell aplasia
b. Anemia of renal failure
c. Anemia of endocrine disease
B. Disturbance of the proliferation and maturation of differentiated stem cells
1. Defective DNA synthesis (megaloblastic anemia):
a. Vitamin B12 deficiency
b. Folic acid deficiency
2. Defective hemoglobin synthesis (hypochromic anemias):
a. Iron deficiency anemia (defective heme synthesis)
b. Thalassemia (defective globin synthesis)
3. Unknown or multiple mechanisms
a. Anemia associated with bone marrow infiltration
b. Anemia of chronic disease
c. Lead poisoning
d. Sideroblastic anemia
III. Increased RBC Destruction (Hemolytic Anemias)
A. Intrinsic Disorders
1. Membrane Defects
a. Hereditary spherocytosis
b. Hereditary ovalocytosis
c. Hereditary acanthocytosis (rare)
d. Hereditary stomatocytosis (rare)
e. Paroxysmal nocturnal hemoglobinuria (PNH)
2. Metabolic Defects
a. Glucose-6-phosphate dehydrogenase deficiency
b. Pyruvate kinase deficiency
3. Hemoglobin Defects
a. Hemoglobinopathies
b. Thalassemia
B. Extrinsic Disorders
1. Chemical Agents
2. Vegetable and Animal Poisons
3. Infectious Agents
4. Physical Agents
a. Heat
b. Traumatic hemolysis (Red Cell Fragmentation Syndrome)
5. Immunologically Caused Hemolytic Anemias
Laboratory Investigation of Anemia
Blood count ‑ red blood cell count
hemoglobin
hematocrit
A. Morphology of red blood cell may yield important clues
1. Size
2. Chromia, or color
3. Poikilocytosis ‑ abnormally shaped cells due to either premature aging or abnormal maturation.
a. Crenated cell ‑ artifact of slide preparation. From use of a hypertonic staining fluid ‑draws water out of the cell.
b. Burr cell ‑ cell has blunt horns seen in anemias associated with chromic kidney diseases i.e. uremia
c. Acanthocytes ‑ due to error of lipoprotein metabolism/rare. Associated with mental retardation.
d. Elliptocytosis ‑ cells oval in shape. May be a genetic defect. Homozygous – causes hemolytic anemia. May also be seen in cases of severe burns
e. Sickle cells ‑ contain abnormal Hgb. Upon oxygen deprivation, the cells form a sickle shape.
f. Howell ‑ Jolly bodies ‑ remnant of DNA or RNA. Commonly seen in splenectomy patients.
g. Cabot rings ‑ similar to Howell Jolly bodies
h. Basophilic stippling ‑ due to poisoning by heavy metals, especially lead. Results from precipitation of ribosomal material within the red cell.
i. Pappenheimer bodies ‑ also called Siderocytes. Can get them in small numbers in all anemias except Fe deficiency. Must be specially stained. Maturation defect.
j. Schistocytes ‑ irregularly shaped fragments of red cells which may be seen in hemolytic anemias.
Polychromasia ‑ (Reticulocytosis)
Polychromatophilic erythrocytes are newly released red cells, lilac in color and contain residual RNA which can be stained with a supravital stain such as New Methylene Blue. Levels of 0. 5‑1.5 % are usually considered normal. A lot of polychromasia indicates increased cell production, usually hemolysis. Lots of new cells have to be produced to keep up with the loss.
Blood Loss Anemia
Posthemorrhagic anemia is an anemia which develops as a result of hemorrhage. There are two types of anemias of this group according to the character of hemorrhage: 1) acute posthemorrhagic and 2) chronic posthemorrhagic anemia.
Acute posthemorrhagic anemia arises after fast massive hemorrhage as a result wounding of vessels or their damage by pathological process.
Chronic posthemorrhagic anemia develops after repeated hemorrhages, caused by injury of blood vessels during number of diseases (dysmenorrhea, ulcer of stomach, hemorrhoids etc.)
The picture of blood of acute posthemorrhagic anemias depends on time which has passed after hemorrhage. Depending on it it is possible pick out three periods, each of them is characterized by the certain picture of peripheral blood.
1. The first several hours after acute hemorrhage. At this period of time the total amount of blood, and also total number of erythrocytes in an organism decreases. However in unit of blood volume the contents of erythrocytes and concentration of hemoglobin is normal.
2. The period of time from several hours untill several days after acute hemorrhage. Dillution of blood takes place as a result of transition of liquid from interstitial spaces into blood vessels. As a result of it the quantity of erythrocytes and hemoglobin in unit of volume of blood decreases, as well as hematocrit. A color index stays without changes (normochromic anemia). Qualitative changes of erythrocytes in blood smear are not found yet.
3. The period of time from several days untill 1-2 weeks after acute hemorrhage. The most typical feature of picture of blood in this period is occurrence of plenty regenerative forms of erythrocytes, due to amplification of erythropoiesis in red bone marrow. Because young unripe erythrocytes contain less hemoglobin in comparison with mature cells, the color index decreases also and anemia becomes hypochromic.
During chronic posthemorrhagic anemia after the loss of iron hematologic attributesof irondeficiency anemia develop: concentration of hemoglobin and color index decrease, in blood smear there are degenerate forms of erythrocytes (micro- and poikilocytosis, hypochromy). Quantity of erythrocytes and hematocrit may remain without changes.
Iron-deficiency anemia
Iron has a pivotal role in many metabolic processes, and the average adult contains 3–5 g of iron, of which two-thirds is in the oxygen carrying molecule haemoglobin.
A normal Western diet provides about 15·mg of iron daily, of which 5–10% is absorbed (~·1·mg), principally in the duodenum and upper jejunum, where the acidic conditions help the absorption of iron in the ferrous form. Absorption is helped by the presence of other reducing substances, such as hydrochloric acid and ascorbic acid. The body has the capacity to increase its iron absorption in the face of increased demand, for example, in pregnancy, lactation, growth spurts
and iron deficiency. Once absorbed from the bowel, iron is transported across the mucosal cell to the blood, where it is carried by the protein transferrin to developing red cells in the bone marrow. Iron stores comprise ferritin, a labile and readily accessible source of iron and haemosiderin, an insoluble form found predominantly in macrophages. About 1·mg of iron a day is shed from the body in urine, faeces, sweat and cells shed from the skin and gastrointestinal tract. Menstrual losses of an additional 20·mg per month and the increased requirements of pregnancy (500–1000·mg) contribute to the higher incidence of iron deficiency in women of reproductive age.

Causes of iron deficiency anaemia
Reproductive system
• Menorrhagia
Gastrointestinal tract
Bleeding
• Oesophagitis
• Oesophageal varices
• Hiatus hernia (ulcerated)
• Peptic ulcer
• Inflammatory bowel disease
• Haemorrhoids (rarely)
• Carcinoma: stomach, colorectal
• Angiodysplasia
• Hereditary haemorrhagic telangiectasia (rare)
Malabsorption
• Coeliac disease
• Atrophic gastritis (also may result from iron deficiency)
Physiological
• Growth spurts (especially in premature infants)
• Pregnancy
Dietary
• Vegans
• Elderly
• Other
• Patients with chronic renal failure undergoing haemodialysis and receiving erythropoietin
Laboratory investigation of iron-deficiency anemia
1. Total blood count.
RBC
Reticulocytes
Platelets
RBC parameters:
· MCV (mean corpuscular volume – N 75-95mcm3)
· MCH (mean corpuscular hemoglobin – N 24-33 picograms),
· MCHC (mean corpuscular hemoglobin concentration – N 30-38 %),
· RDW (red cell distribution width – N 11,5-14 %).
At the onset of disease amount of red blood cells is not reduced, but they are reduced in size (microcytes) and not saturated enough with hemoglobin (hypochromiya). Color index is low (0,7-0,5) and MCHC also decreases. In blood smears small hypochromic erythrocytes are dominated with unequal size and shape (anizocytosis, poykilocytosis). In severe anemia erythroblasts may appear. The number of reticulocytes does not change. But if the anemia is caused by acute hemorrhage, immediately after it, the level of reticulocytes is increased, which is an important sign of bleeding. Osmotic resistance of erythrocytes is not changed or is slightly increased. The level of platelets does not change, only in case of hemorrhage is slightly increased.
2. Biochemical profile.
Ferritin, serum
Ferritin is a high molecular weight protein that consists of approximately 20 % iron. It is found in all cells, but especially in hepatocytes and reticuloendothelial cells, where it serves as an iron reserve. A small amount is present in plasma and serum and reflects iron stores in normal individuals. Iron is released from ferritin and binds to transferrin for transport to developing red blood cells in the bone marrow. Inadequate iron stores may result in iron deficient erythropoiesis.
Ferritin is also a useful screening test to distinguish iron deficiency from thalassemia minor in patients with anemia and erythrocyte microcytosis; ferritin is decreased in iron deficiency and normal or increased in thalassemia.
Interpretation
Reference Range: 45-340 ng/dL
· Values less than 100 ng/dL usually indicate depleted iron stores
· Ferritin is decreased in patients with an iron-depletion state
· Ferritin may be normal to mildly increased in functional iron deficiency
(anemia of inflammation) and in anemia secondary to thalassemia minor
· Ferritin is increased in inflammation
· Ferritin may be markedly increased in hereditary hemochromatosis and
other iron overload states, acute hepatitis, and many malignancies and in Gaucher’s disease
Iron, serum
Ingested iron is absorbed primarily from the intestinal tract, temporarily stored as ferritin in mucosal cells, and then released into the blood as Fe3+ - transferrin in equilibrium with a very small amount of free Fe3+. Serum iron can be used as one test to evaluate patients for iron deficiency, especially in combination with iron binding capacity (transferrin and transferrin saturation). Serum iron alone is unreliable due to considerable physiologic variation in the results.
Many normal subjects demonstrate a predictable diurnal variation with highest values in the morning and lowest values in the evening. Values in an individual may vary 10-40 % within a single day or day-to-day due to changes in iron absorption, marrow iron uptake, or storage iron outflow. Therefore, serum iron results should always be interpreted in the context of other studies.
Interpretation
Reference Range: 50-150 ug/dL
· Useful for diagnosis of iron depletion states especially when used in combination with transferrin and transferrin saturation
· Can be used for evaluation of chronic iron overload states
· Subject to physiologic variation including diurnal variation, and variation in response to iron therapy
Iron binding capacity, total (total serum transferrin) N- 1,7-4,7 mg/l, or 30,6 -84,6 micromole/l
Soluble transferrin receptor (sTfR), serum
Transferrin receptors are present on the external surface of the plasma membrane. In order for iron to be internalized into cells, the iron-transferrin complex binds to these receptors. It is then internalized through endosomes and the iron released into the cytoplasm. Proteolytic cleavage of the transferrin receptor releases a truncated version of the transferrin receptor as soluble transferrin receptor circulating in the blood. Membrane expression of transferrin receptors (TfR) are regulated by iron status. There is increased expression of TfR in iron deficiency states and this results in an increase in soluble TfR as well. In iron repletion states, there is a decrease in membrane and soluble TfR. TfR is not an acute phase reactant. While ferritin, which is an acute phase reactant, increases in response to inflammatory states, malignancy, infection, and chronic disease, soluble TfR is not affected by these confounding pathologies and may help determine the status of iron stores in patients with inflammation. TfR should not be used routinely for evaluation of iron status as it is referral test for most hospital laboratories with a higher cost and slower turn around time than ferritin, transferrin and transferrin saturation.
Interpretation
Reference Range: 1.8 – 4.6 mg/L
· Useful for evaluating iron status in patients with inflammation
· sTfR concentration is inversely related to iron status; sTfR elevates in
response to iron deficiency and decreases in response to iron repletion
· Patients with hemolysis or recent blood loss may have falsely elevated
sTfR levels
· sTfR is elevated in patients with thalassemia and sickle cell disease.
Transferrin, serum
Transferrin is the principle plasma protein for transport of iron. Its concentration
correlates with the total iron-binding capacity of serum. For diagnosis of iron depletion states, transferrin and iron-binding capacity may be used interchangeably. Transferrin is synthesized primarily in the liver. In otherwise healthy individuals with iron depletion states, transferrin levels in serum increase due to an increase in synthesis. High levels can be seen in pregnancy and during estrogen administration. Decreased transferrin may be seen in chronic liver disease, malnutrition, and protein loss. It is important to note that transferrin is decreased in malignancy and in both acute and chronic inflammation.
Interpretation
Reference Range: 200-400 mg/dL
· Transferrin is significantly increased in iron depletion states
· Transferrin is decreased in inflammatory states including anemia of chronic inflammation (functional iron deficiency)
· May be decreased in malnutrition, chronic liver disease, malignancy, and protein loss
· High levels of transferrin may be seen in pregnancy (and may be an indicator of iron depletion) and during estrogen administration.
Transferrin Saturation
Transferrin, the principal plasma protein for transport of iron, binds iron strongly at
physiologic pH. Transferrin is generally 20-45% saturated with iron. The additional amount of iron that can be bound is the unsaturated iron-binding capacity (UIBC). The sum of the serum iron and UBIC represents the total iron-binding capacity (TIBC). TIBC is an indirect measure of transferrin concentration and the two terms are often used interchangeably. The transferrin saturation (TSat) is usually reported as percent saturation (100 x serum iron/TIBC or transferrin).
Interpretation
Reference Range: 20-45 %
·Transferrin saturation less than 20 % is indicative of an iron deficiency state, either, latent iron deficiency, functional iron deficiency (usually associated with a decrease in transferrin but a disproportionately larger decrease in iron resulting in a transferrin saturation < 20 %) or true iron deficiency where a decrease in serum iron is associated with an increase in transferrin
· Transferrin saturation > 45 % may suggest hereditary hemochromatosis
· Transferrin saturation > 45 % may indicate an iron overload state (hemosiderosis) due to multiple transfusions or iron poisoning
· A transient increase in transferrin saturation is seen after intravenous iron infusion. The duration is dependent on the type and dose of the iron infusion.
3. Investigation of bone marrow.
Diagnostic bone marrow sampling is seldom performed in simple iron defi ciency, but, if the diagnosis is in doubt, a marrow aspirate may be carried out to demonstrate absent bone marrow stores.
4. Investigation of free protoporphyrin in erythrocytes.
Porphyrins are widely distributed in living cells throughout nature and play essential roles in various metabolic processes such as photosynthesis, transportation of oxygen and cellular respiration. In man the most important of the porphyrins is protoporphyrin 9, type III, which in combination with iron and specific proteins forms such compounds as hemoglobin, myoglobin, cytochrome,
peroxidase and catalase. In addition to its occurrence in these compounds, protoporphyrin has been found in apparently uncombined or free form in erythrocytes, hence the term "free erythrocyte protoporphyrin"
Adults: 16-36 mcg/dL red cells (0.28-0.64 micromol/L red cells)
The sequence of events (left to right) that occur with gradual depletion of body stores of iron. Serum ferritin and stainable iron in tissue stores are the most sensitive laboratory indicators of mild iron deficiency and are particularly useful in differentiating iron deficiency from the anemia of chronic disorders. The percentage saturation of transferrin with iron and free erythrocyte protoporphyrin values do not become abnormal until tissue stores are depleted of iron. Subsequently, a decrease in the hemoglobin concentration occurs because iron is unavailable for heme synthesis. Red blood cell indices do not become abnormal for several months after tissue stores are depleted of iron.
Normal blood smear: RBC, platelets, lymphocyte
Hemolytic anemias
Red cells being enucleated, their survival within the blood circulation depends on the stability of their various components that cannot be replaced through protein synthesis. The normal life span of the red cell is 120 days. The normal red blood cell production compensates for the destruction and loss of red cells. Thus one in every 120 red cells (or 0.8%) is replaced every day by a newly formed red cell. These newly formed red cells, called "reticulocytes", can be recognized with
special stains because of the transient persistence of ribosomes in their cytoplasm.
Hemolysis is defined as a shortening of the red cell survival below the normal of 120 days.
MECHANISMS OF HEMOLYSIS
1. Fragmentation:
Mechanical destruction of the red cells. Examples include red cells trapped by malfunctioning artificial heart valves or fragmentation of red cells that are going through fibrin clots in the microcirculation (e.g. disseminated intra-vascular coagulation).
2. Osmotic Lysis:
The concentration of protein in the red cell is very high (32-36 gm% of hemoglobin). This creates a high osmotic force that tends to let water and sodium "leak" into the cell. This water needs to be pumped out by a membrane pump that uses ATP as a power source. If not enough ATP can be generated (as in pyruvate kinase deficiency) then the pump does not function properly and the red cell swells with water and may ultimately explode.
3. Erythrophagocytosis:
In autoimmune hemolytic anemia, red cell coated with antibodies are taken up and destroyed by macrophages, which have receptors for the Fc portion of the antibody (immunoglobulin).
4. Complement – Mediated Hemolysis:
The "complement" system is made up of a series of serum proteins which are sequentially activated, ultimately creating an active enzyme that punches a hole in the membrane of the red cell. This results in water rushing into the red cell, thus causing osmotic lysis. The complement system may be activated by bacteria, viruses, and certain antibodies.
LABORATORY FINDINGS IN HEMOLYSIS
1. The blood smear should be examined for potential clues about the cause of hemolysis. The reticulocyte count is elevated, indicating the increased production of red cells in an attempt to compensate for abnormal red cell losses.
2. Biochemical changes indicate the increased turnover of red cells:
· increased serum bilirubin (unconjugated form, or "indirect")
· increased serum lactate dehydrogenase (L.D.H.), an enzyme released from red cells
· increased serum iron (due to its release from the heme moiety of the hemoglobin)
In intravascular hemolysis:
· Decreased serum haptoglobin
· Increased free serum hemoglobin
· Hemosiderinuria
3. The bone marrow is sometimes examined to clarify the diagnosis. It will then show an expansion (hyperplasia) of the erythroid precursors (decreased marrow fat and lower myeloid/erythroid ratio as the erythroid cells increase in number to compensate for the hemolysis).
Rarely, the red cell survival is directly evaluated using autologous red cells labeled with radioactive chromium-51 (51Cr).
Hemoglobinopathy: A genetic defect that results in abnormal structure of one of the globin chains of the hemoglobin molecule.
Thalassemia: A genetic defect that results in production of an abnormally low quantity of a given hemoglobin chain or chains. The defect may affect the a, b, g, or d chain, or may affect some combination of the b, g, and d chain in the same patient (but never the a and b chain together). The result is an imbalance in production of globin chains and the production of an inadequate number of red cells.
The Hb S gene is found primarily in populations of native tropical African origin (which include most African-Americans). The incidence of the gene in some African populations is as high as 40 %; in African-Americans the incidence is 8 %. Unfortunately, homozygous expression produces sickle cell disease, which is a chronic hemolytic anemia and vasoocclusive condition that usually takes the life of the patient.
Sickle cell anemia is a particularly bad disease in that not only is it a hemolytic anemia, but also a vasoocclusive condition. The clinical findings can then be divided into one of these two groups:
a. Effects of chronic hemolysis
1. Anemia. Pretty much self-explanatory
2. Jaundice, due to rapid heme turnover and subsequent generation of bilirubin
3. Cholelithiasis. It has been classically taught that sickle cell patients are prone to the formation of calcium bilirubinate gallstones due to excess bilirubin secretion into the hepatobiliary tree.
4. Aplastic crisis. Many of us have brief episodes of marrow aplasia as a result of common viral infections. With a normal erythrocyte life span of 120 days, no anemia results from an unnoticed marrow shut-down of a few days. However, the sickle cell patients, with their markedly abbreviated RBC life span, can have a precipitous fall in hematocrit (and retic count) under such conditions. This may be life-threatening.
5. Hemolytic crisis. Most sickle cell patients establish a stable, tonic level of hemolysis. Rarely, for obscure reasons, they experience a catastrophic fall in hematocrit, increasing intensity of jaundice, and increasing reticulocyte count. This is called a “hemolytic crisis.”
b. Effects of vaso-occlusion
1. Dactylitis. Resulting presumably from infarction or ischemia of the bones of the hands and feet, this is often the presenting manifestation of sickle cell disease in a six-months-old infant. The hands and feet are swollen and painful.
2. Autosplenectomy. In childhood, the spleen is enlarged due to excess activity in destruction of the sickled erythrocytes. Gradually, the spleen infarcts itself down to a fibrous nubbin.
3. Priapism. This refers to a painful and sustained penile erection, apparently due to sludging of sickled cells in the corpora cavernosa. Sometimes the penis has to be surgically decompressed. Repeated episodes of priapism cause the spongy erectile tissues to be replaced by fibrous tissue, with impotence being the end result.
4. Renal papillary necrosis. The physiologic function of the loops of Henle make the renal medulla an eldritch, unbodylike area of high hematocrit, high osmolarity, low pH, hemodynamic stasis, and low pO2. All of these conditions predispose to sickling and infarctive loss of the papillae of the pyramids. The result is inability to concentrate and dilute urine. Even sickle cell trait individuals may experience episodes of hematuria, presumably due to this mechanism.
5. Infarctive (painful) crisis. Increased sickling activity may be brought about by any general stress on the body, especially infection. Almost any organ may suffer acute infarction (includinmg the heart), and pain is the chief symptom.
6. Sequestration crisis. This occurs mostly in infants and young children and is characterized by sudden pooling of sickled erythrocytes in the RES and vascular compartment. This produces a sudden fall in hematocrit. Sequestration crisis may be the most common cause of death in sickle cell patients in the youngest age group.
7. Leg ulcers. After all of the disasters mentioned above, this seems trivial. However, the deep, nonhealing ulcers of skin and tela subcutanea (classically around the medial malleolus) may be the only clinical manifestation of sickle cell disease in an otherwise well-compensated patient. These may be the only bugaboo standing between the patient and a productive, financially solvent life.
Megaloblastic anemia
Megaloblastic anemia is an anemia (of macrocytic classification) that results from inhibition of DNA synthesis in red blood cell production. When DNA synthesis is impaired, the cell cycle cannot progress from the growth stage to the mitosis stage. This leads to continuing cell growth without division, which presents as macrocytosis.
Causes:
1.Vitamin B12 deficiency leading to folate deficiency:
§ Achlorhydria-induced malabsorption
§ Deficient intake
§ Deficient intrinsic factor (pernicious anemia or gastrectomy
)
§ Selective Vitamin B12 malabsorption (congenital—juvenile megaloblastic anemia 1—and drug-induced)
§ Nitrous oxide anesthesia (usually requires repeated instances).
§ Increased needs: pregnancy, infant, rapid cellular proliferation, and cirrhosis
§ Malabsorption (congenital and drug-induced)
§ Intestinal and jejunal resection
§ (indirect) Deficient thiamine and factors (e.g., enzymes) responsible for folate metabolism.
3. Combined Deficiency: Vitamin B12 & folate.
§ Inherited Pyrimidine Synthesis Disorders: Orotic aciduria
§ Inherited DNA Synthesis Disorders
a. Folic acid antagonists (methotrexate)
b. Purine synthesis antagonists (6-mercaptopurine)
c. Pyrimidine antagonists (cytosine arabinoside)
d. Phenytoin
The blood film can point towards vitamin deficiency:
§ Decreased red blood cell (RBC) count and hemoglobin levels
§ Increased mean corpuscular volume (MCV, >95 fl) and mean corpuscular hemoglobin (MCH)
§ Normal mean corpuscular hemoglobin concentration (MCHC, 32–36 g/dL)
§ The platelet count may be reduced.
§ Anisocytosis (increased variation in RBC size) and poikilocytosis (abnormally shaped RBCs).
§ Macrocytes (larger than normal RBCs) are present.
§ Ovalocytes (oval-shaped RBCs) are present.
§ Howell-Jolly bodies (chromosomal remnant) also present.
Blood chemistries will also show:
§ Increased homocysteine and methylmalonic acid in Vitamin B12 deficiency
§ Increased homocysteine in folate deficiency
Bone marrow shows megaloblastic hyperplasia.
§ Long-term use of ranitidine hydrochloride may contribute to deficiency of vitamin B12.
§ The diabetes medication metformin may interfere with B12 dietary absorption.
§ Hereditary causes such as severe MTHFR deficiency, homocystinuria, and transcobalamin deficiency.
3. Decrease or abolishment of deep muscle-tendon reflexes
4. Pathological reflexes — Babinski, Rossolimo and others, also severe paresis
Myelodysplastic syndrome (MDS)
MDS are a morphologically and clinically heterogeneous group of acquired stem cell disorders characterized by ineffective and dysplastic haematopoiesis in one or more cell lines that affect predominantly older subjects. The patients usually present with anaemia and dysplastic morphologic abnormalities in cells of more than one lineage (neutropenia, thrombocytopenia, monocytosis, or a combination of types of cytopenias). The diagnosis is essentially based on the morphological examination of a blood smear and a bone marrow biopsy specimen. The French-American-British classification divides the MDS into five subtypes based on the number of immature blast cells and morphological abnormalities in blood and bone marrow and the presence of ringed sideroblasts.
§ Neutropenia (low neutrophil count) —increased susceptibility to infection
§ splenomegaly or rarely hepatomegaly;
§ abnormal granules in cells, abnormal nuclear shape and size; and/or
§ chromosomal abnormalities, including chromosomal translocations and abnormal chromosome number.
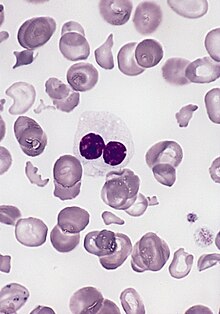
Erythrocyte with a delicate Cabot ring (arrow) (here in a case of osteomyelosclerosis)
Fixation and staining artifacts.
CLINICAL LABORATORY DIAGNOSTICS OF HEMOBLASTOSIS
Etiology is Unknown: Possible Causes:
B. Marrow damage due to irradiation increases the frequency of some leukemias, but not others.
D. Possible genetic factors have been implicated, especially in Chronic Lymphocytic Leukemia.
2. If the prominent cell line is of the lymphoid series it is a lymphocytic leukemia
Therefore, there are four basic types of leukemia.
II. Acute lymphocytic leukemia – ALL - (includes T cell, B cell, and Null cell)
III. Chronic myelocytic leukemia – CML - (includes myelocytic and myelomonocytic)
Acute leukemias can occur in all age groups
n ALL is more common in children
n AML is more common in adults
Chronic leukemias are usually a disease of adults
n CLL is extremely rare in children and unusual before the age of 40
Age Distribution: optimum ages for development:
5. Monocytic: middle age (rarely before 30)
Comparison of acute and chronic leukemias:
Clinical onset sudden insidious
Course (untreated) 6 mo. or less 2-6 years
Leukemic cells immature >30% blasts more mature cells
Thrombocytopenia prominent mild
Lymphadenopathy mild present; often prominent
Splenomegaly mild present; often prominent
Malignant transformation of a stem cell leading to unregulated proliferation and
Pathophysiology of the clinical manifestations of acute leukemias
1. Marrow failure due to infiltration
-thrombocytopenia -bleeding, spontaneous bruising
-neutropenia-infections, sepsis
2. Infiltration of other organs
-liver, spleen, lymph nodes (particularly in ALL)
-gums –gum hypertrophy (monocytic subtype of AML)
-bone pain, esp. in children with ALL
3. Leukostasis (only seen with WBC >>50x109 /L)
-lungs -pulmonary infiltrates, hypoxemia
-exposure of substances that can initiate coagulation can cause DIC
The lab diagnosis is based on two things:
2. Identification of the cell lineage of the leukemic cells
Anemia (normochromic, normocytic)
The degree of peripheral blood involvement determines classification:
n Leukemic – increased WBCs due to blasts
n Subleukemic – blasts without increased WBCs
n Aleukemic – decreased WBCs with no blasts
Classification of the immature cells involved may be done by:
Myeloblast with auer rods Lymphoblasts
Auer rods in AML
Cytochemistry
Cytochemistry – help to classify the lineage of a leukemic cell (myeloid versus lymphoid). Myeloperoxidase – is found in the primary granules of granulocytic cells starting at the late blast stage. Monocytes may be weakly positive.
Sudan black stains phospholipids, neutral fats and sterols found in primary and secondary granules of granulocytic cells and to a lesser extent in monocytic lysosomes.
Rare positives occur in lymphoid cells. Nonspecific esterase – is used to identify monocytic cells which are diffusely positive. T lymphocytes may have focal staining.
Acid phosphatase may be found in myeloblasts and lymphoblasts. T lymphocytes have a high level of acid phosphatase and this can be used to help make a diagnosis of acute T-lymphocytic leukemia.
Leukocyte alkaline phosphatase – is located in the tertiary granules of segmented neutrophils, bands and metamyelocytes. The LAP score is determined by counting 100 mature neutrophils and bands. Each cell is graded from 0 to 5. The total LAP score is calculated by adding up the scores for each cell.
Chronic Lymphocytic Leukemia
Neoplastic proliferation of small mature-appearing lymphocytes
>99 % are B-cell derived
Older patients (> 40)
Disease may be discovered incidentally
Fatigue, weakness, weight loss, anorexia, and/or recurrent infections may occur
Variable splenomegaly and non tender lymphadenopathy
Leukemic counterpart of small lymphocytic
Lymphoma
Lab diagnostics
Absolute lymphocytosis (> 4000/mm3)
Smudge cells
Variable anemia, neutropenia, and thrombocytopenia
Hypercellular bone marrow with lymphocytic infiltrates
Hypogammaglobulinemia
Elevated serum LDH
Bone Marrow in CLL
Chronic Myelocytic Leukemia (CML)
15-20 % of all leukemias
Young or middle-aged patients
Asymptomatic, fatigue, abdominal fullness, early satiety, weight loss, anorexia
Splenomegaly, bone pain, bone tenderness
Proliferation of myeloid cells in blood and bone marrow (mostly myelocytes polys)
Slow progression (3 yr survival without Rx)
Ph 1 chromosome in > 95 %
Peripheral Blood in CML
Bone Marrow in CML
Hypercellular bone marrow
High Myeloid : Erythroid Ratio
Myeloid hyperplasia
Relatively few blast cells
Mostly mature neutrophils
Increased basophils and eosinophils
Increased megakaryocytes
Bone Marrow in CML
Multiple Myeloma
Multiple myeloma, also known as plasma cell myeloma or Kahler's disease, is a cancer of plasma cells, a type of white blood cell normally responsible for producing antibodies. In multiple myeloma, collections of abnormal plasma cells accumulate in the bone marrow, where they interfere with the production of normal blood cells. Most cases of myeloma also feature the production of a paraprotein — an abnormal antibody which can cause kidney problems. Bone lesions and hypercalcemia (high calcium levels) are also often encountered.
Myeloma is diagnosed with blood tests (serum protein electrophoresis, serum free kappa/lambda light chain assay), bone marrow examination, urine protein electrophoresis, and X-rays of commonly involved bones.
The presence of unexplained anemia, kidney dysfunction, a high erythrocyte sedimentation rate (ESR), lytic bone lesions, elevated beta-2 microglobulin, and/or a high serum protein (especially raised globulins or immunoglobulin) may prompt further testing. The globulin level may be normal in established disease. A doctor will request protein electrophoresis of the blood and urine, which might show the presence of a paraprotein (monoclonal protein, or M protein) band, with or without reduction of the other (normal) immunoglobulins. One type of paraprotein is the Bence Jones protein which is a urinary paraprotein composed of free light chains. Quantitative measurements of the paraprotein are necessary to establish a diagnosis and to monitor the disease. The paraprotein is an abnormal immunoglobulin produced by the tumor clone. Very rarely, the myeloma is nonsecretory (not producing immunoglobulins).
In theory, multiple myeloma can produce all classes of immunoglobulin, but IgG paraproteins are most common, followed by IgA and IgM. IgD and IgE myeloma are very rare. In addition, light and or heavy chains (the building blocks of antibodies) may be secreted in isolation: κ- or λ-light chains or any of the five types of heavy chains (α-, γ-, δ-, ε- or μ-heavy chains).
Additional findings include: a raised calcium (when osteoclasts are breaking down bone, releasing calcium into the bloodstream), raised serum creatinine due to reduced renal function, which is mainly due to casts of paraprotein deposition in the kidney, although the cast may also contain complete immunoglobulins and albumin.
Cancer of plasma cells
Disease of older men and women (> 60 years)
Produce abundant useless monoclonal Ig (paraprotein, M-protein)
Decreased normal Ig, infections occur
Lytic bone lesions, bone pain, pathologic fractures, hypercalcemia
Tumorous masses of plasma cells (spine, skull, ribs, pelvis)
Renal failure may develop
Peripheral Blood in MM
Bone Marrow in Multple Myeloma
Lymphogranulomatosis
Lymphogranulomatosis – is a malignant tumor of lymphoid tissue with lesion of lymph nodes and organs, characterized by growth of the giant cells called Reed-Sternberg- Beresovsky cells, large and small atypical cells (Hodgkin,s cells) and inflammatory infiltration.
Hodgkin disease: a giant mononuclear cell with a large nucleolus (arrow) and wide cytoplasmic
layer (Hodgkin cell), surrounded by small and medium-sized lymphocytes.
Hodgkin disease: giant binuclear cell (Reed–Sternberg giant cell).
Hodgkin's lymphoma, also known as Hodgkin lymphoma and previously known as Hodgkin's disease, is a type of lymphoma, which is a cancer originating from white blood cells called lymphocytes.
Patients with Hodgkin's lymphoma may present with the following symptoms:
§ Lymph nodes: the most common symptom of Hodgkin's is the painless enlargement of one or more lymph nodes, or lymphadenopathy. The nodes may also feel rubbery and swollen when examined. The nodes of the neck and shoulders (cervical and supraclavicular) are most frequently involved (80–90 % of the time, on average). The lymph nodes of the chest are often affected, and these may be noticed on a chest radiograph.
§ Unexplained weight loss
§ Splenomegaly: enlargement of the spleen occurs in about 30 % of people with Hodgkin's lymphoma. The enlargement, however, is seldom massive and the size of the spleen may fluctuate during the course of treatment.
§ Hepatomegaly: enlargement of the liver, due to liver involvement, is present in about 5 % of cases.
§ Hepatosplenomegaly: the enlargement of both the liver and spleen caused by the same disease.
§ Red-coloured patches on the skin, easy bleeding and petechiae
due to low platelet count (as a result of bone marrow infiltration, increased trapping in the spleen etc. – i.e. decreased production, increased removal)
the syringe. The syringe body is separated from the needle and the mandrel
Cell composition in the bone marrow: normal values (%)
|
Cell densities vary widely, 0.5–2 per view field during screening at low magnification. |
|||
Squash preparation and meandering smear for the cytological analysis of bone marrow spicules
Bone Marrow: Medullary Stroma Cells