Investigation of acid-base state of blood and respiratory function of
erythrocytes. Pathological
forms of hemoglobin.
Investigation of blood plasma proteins of
inflammation acute phase, own and indicator enzymes. Non-protein
nitrogenous containing and nitrogen not containing organic components of blood.
residual nitrogen
Blood is a liquid tissue. Suspended in the watery plasma are seven types of cells and cell fragments.
·
red blood cells (RBCs)
or erythrocytes
·
platelets or thrombocytes
·
five kinds of white blood cells
(WBCs) or leukocytes
o
Three kinds of granulocytes
§
neutrophils
§
eosinophils
§
basophils
o
Two kinds of leukocytes without
granules in their cytoplasm
§
lymphocytes
§
monocytes
|
If
one takes a sample of blood, treats it with an agent to prevent clotting, and
spins it in a centrifuge,
·
the red cells settle to the bottom
·
the white cells settle on top of
them forming the "buffy coat".
The
fraction occupied by the red cells is called the hematocrit. Normally
it is approximately 45%. Values much lower than this are a sign of anemia.
|

|
Biological functions of the blood
The blood is the most specialized fluid tissue which
circulates in vascular system and together with lymph and intercellular space compounds
an internal environment of an organism.
The blood executes such functions:
1. Transport of gases –
oxygen from lungs is carried to tissues and carbon dioxide from tissues to
lungs.
2. Transport of nutrients to all cells of
organism (glucose, amino acids, fatty acids, vitamins, ketone bodies, trace
substances and others). Substances such as urea, uric acid, bilirubin and
creatinine are taken away from the different organs for ultimate excretion.
3. Regulatory or hormonal function – hormones
are secreted in to blood and they are transported by blood to their target
cells.
4. Thermoregulation
function - an exchange of heat between tissues and blood.
5. Osmotic function-
sustains osmotic pressure in vessels.
6. Protective function- by
the phagocytic action of leucocytes and by the actions of antibodies, the blood
provides the most important defense mechanism.
7. Detoxification function
- neutralization of toxic substances which is connected with their
decomposition by the help of blood enzymes.
Blood
performs two major functions:
·
transport through the body of
o
oxygen and carbon dioxide
o
food molecules (glucose, lipids,
amino acids)
o
ions (e.g., Na+, Ca2+,
HCO3−)
o
wastes (e.g., urea)
o
hormones
o
heat
·
defense of the body against
infections and other foreign materials. All the WBCs participate in these
defenses.
The formation of blood cells (cell types and acronyms are
defined below)
All the
various types of blood cells
·
are produced in the bone marrow
(some 1011 of them each day in an adult human!).
·
arise from a single type of cell
called a hematopoietic stem cell — an "adult" multipotent stem cell.
These stem cells
·
are very rare (only about one in
10,000 bone marrow cells);
·
are attached (probably by adherens junctions)
to osteoblasts
lining the inner surface of bone cavities;
·
express a cell-surface protein
designated CD34;
·
produce, by mitosis, two kinds of
progeny:
o
more stem cells (A mouse that has had
all its blood stem cells killed by a lethal dose of radiation can be saved by
the injection of a single living stem cell!).
o
cells that begin to differentiate
along the paths leading to the various kinds of blood cells.
Which
path is taken is regulated by
·
the need for more of that type of
blood cell which is, in turn, controlled by appropriate cytokines
and/or hormones.
Examples:
·
Interleukin-7 (IL-7)
is the major cytokine in stimulating bone marrow stem cells to start down the
path leading to the various lymphocytes
(mostly B cells
and T cells).
·
Erythropoietin (EPO),
produced by the kidneys, enhances the production of red blood cells
(RBCs).
·
Thrombopoietin (TPO),
assisted by Interleukin-11 (IL-11), stimulates the production of megakaryocytes.
Their fragmentation produces platelets.
·
Granulocyte-macrophage
colony-stimulating factor (GM-CSF), as its name
suggests, sends cells down the path leading to both those cell types. In due
course, one path or the other is taken.
o
Under the influence of granulocyte
colony-stimulating factor (G-CSF), they differentiate into neutrophils.
o
Further stimulated by interleukin-5 (IL-5)
they develop into eosinophils.
o
Interleukin-3 (IL-3)
participates in the differentiation of most of the white blood cells but plays a
particularly prominent role in the formation of basophils
(responsible for some allergies).
o
Stimulated by macrophage
colony-stimulating factor (M-CSF) the granulocyte/macrophage
progenitor cells differentiate into monocytes, macrophages,
and dendritic cells
(DCs).
Biological chemistry of blood cells
Two types of blood cells can be distinguished
- white and red blood cells. White blood cells are called leucocytes. Their
quantity in adult is 4-9 x 109/L.
Red blood cells
are called erythrocytes. Their quantity
in peripheral blood is 4,5-5 x 1012/L.
Besides that, there are also thrombocytes or platelets in blood.
White Blood Cells (leukocytes)
Leucocytes
(white blood cells) protect an organism from
microorganisms, viruses and foreign substances, that provides the immune status
of an organism.
·
are much less numerous than red (the
ratio between the two is around 1:700),
·
have nuclei,
·
participate in protecting the body
from infection,
·
consist of lymphocytes and monocytes
with relatively clear cytoplasm, and three types of granulocytes, whose
cytoplasm is filled with granules.
Leucocytes are divided into
two groups: Granulocytes and agranulocytes. Granulocytes consist of
neutrophils, eosinophils and basophils. Agranulocytes consist of monocytes and
lymphocytes.
http://www.youtube.com/watch?v=8ytkFqAMoa8
http://www.youtube.com/watch?v=ce0Xndms1bc
Neutrophils
Neutrophils comprise of
60-70 % from all leucocytes. Their main function is to protect organisms from
microorganisms and viruses. Neutrophils have segmented nucleus, endoplasmic
reticulum (underdeveloped) which does not contain ribosomes, insufficient
amount of mitochondria, well-developed Golgi apparatus and hundreds of
different vesicles which contain peroxidases and hydrolases. Optimum condition
for their activity is acidic pH. There are also small vesicles which contain
alkaline phosphatases, lysozymes, lactopherins and proteins of cationic origin.
Glucose is the main source
of energy for neutrophils. It is directly utilized or converted into glycogen.
90 % of energy is formed in glycolysis, a small amount of glucose is converted
in pentosophosphate pathway. Activation of proteolysis during phagocytosis as
well as reduction of phosphatidic acid and phosphoglycerols are also observed.
The englobement is accompanied by intensifying of a glycolysis and
pentosophosphate pathway. But especially intensity of absorption of oxygen for
neutrophils - so-called flashout of respiration grows. Absorbed oxygen is spent
for formation of its fissile forms that is carried out with participation
enzymes:
1. NADP*Н -OXYDASE catalyzes formation of super oxide
anion
2. An enzyme
NADH- OXYDASE is responsible for formation
of hydrogen peroxide
3. Мyeloperoxydase catalyzes
formation of hypochloric acid from chloride and hydrogen peroxide
Neutrophils
are motile phagocyte cells that play a key role in acute inflammation. When
bacteria enter tissues, a number of phenomena occur that are collectively known
as acute inflammatory response. When neutrophils and other phagocyte cells
engulf bacteria, they exhibit a rapid increase in oxygen consumption known as
the respiratory burst. This phenomenon reflects the rapid utilization of oxygen
(following a lag of 15-60 seconds) and production from it of large amounts of
reactive derivates, such as O2-, H2O2,
OH. and OCl-
(hypochlorite ion). Some of these products are potent microbicidal
agents. The electron transport chain system responsible for the respiratory
burst contains several components, including a flavoprotein NADPH:O2-oxidoreductase
(often called NADPH-oxidase) and a b-type cytochrome.
The most
abundant of the WBCs. This photomicrograph shows a single neutrophil surrounded
by red blood cells.
Neutrophils
squeeze through the capillary walls and into infected tissue where they kill
the invaders (e.g., bacteria) and then engulf the remnants by phagocytosis.
This
is a never-ending task, even in healthy people: Our throat, nasal passages, and
colon harbor vast numbers of bacteria. Most of these are commensals,
and do us no harm. But that is because neutrophils keep them in check.
However,
·
heavy doses of radiation
·
chemotherapy
·
and many other forms of stress
can
reduce the numbers of neutrophils so that formerly harmless bacteria begin to proliferate.
The resulting opportunistic infection can be life-threatening.
http://www.youtube.com/watch?v=EpC6G_DGqkI&feature=related
Some
important enzymes and proteins of neutrophilis.
Myeloperoxidase
(MPO). Catalyzed following reaction:
H2O2
+ X-(halide) + H+®
HOX + H2O (where X- = Cl-, Br-, I-
or SCN-; HOX=hypochlorous acid)
HOCl,
the active ingredient of household liquid bleach, is a powerful oxidant and is
highly microbicidial. When applied to normal tissues, its potential for causing
damage is diminished because it reacts with primary or secondary amines present
in neutrophils and tissues to produce various nitrogen-chlorine (N-Cl)
derivates; these chloramines are also oxidants, although less powerful than
HOCl, and act as microbicidial agents (eg, in sterilizing wounds) without
causing tissue damage. Responsible for the green color of pus.
NADPH-oxidase.
2O2
+ NADPH ®
2O2- + NADP + H+
Key
component of the respiratory burst. Deficiency may be observed in chronic
granulomatous disease.
Lysozyme.
Hydrolyzes
link between N-acetylmuramic acid and N-acetyl-D-glucosamine found in certain
bacterial cell walls. Abundant in macrophages.
Defensins.
Basic
antibiotic peptides of 29-33 amino acids. Apparently kill bacteria by causing
membrane damage.
Lactoferrin.
Iron-binding
protein. May inhibit growth of certain bacteria by binding iron and may be
involved in regulation of proliferation of myeloid cells.
Neutrophils
contain a number of proteinases (elastase, collagenase, gelatinase, cathepsin
G, plasminogen activator) that can hydrolyze elastin, various types of
collagens, and other proteins present in the extracellular matrix. Such
enzymatic action, if allowed to proceed unopposed, can result in serious damage
to tissues. Most of these proteinases are lysosomal enzymes and exist mainly as
inactive precursors in normal neutrophils. Small amounts of these enzymes are
released into normal tissues, with the amounts increasing markedly during
inflammation. The activities of elastase and other proteinases are normally
kept in check by a number of antiproteinases (a1-Antiproteinase,
a2-Macroglobulin,
Secretory leukoproteinase inhibitor, a1-Antichymotrypsin,
Plasminogen activator inhibitor-1, Tissue inhibitor of metalloproteinase)
present in plasma and the extracellular fluid.
Basophiles
Basophiles make up 1-5% of all blood leukocytes. They
are actively formed in the bone marrow
during allergy. Basophiles take part in
the allergic reactions, in the blood coagulation and intravascular
lipolysis. They have the protein synthesis mechanism, which works due to the
biological oxidation energy . They synthesize the mediators of allergic
reactions – histamine and serotonin, which during allergy cause local
inflammation. Heparin, which is formed in the basophiles, prevents the blood
coagulation and activates intravascular lipoprotein lipase, which splits triacylglycerin.
The
number of basophils also increases during infection. Basophils leave the blood
and accumulate at the site of infection or other inflammation. There they
discharge the contents of their granules, releasing a variety of mediators such
as:
·
histamine
·
serotonin
·
prostaglandins
and leukotrienes
which
increase the blood flow to the area and in other ways add to the inflammatory
process. The mediators released by basophils also play an important part in
some allergic responses such as
·
hay fever and
·
an anaphylactic response
to insect stings.
Eosinophiles
They make up 3-6% of all leukocytes. Eosinophiles as
well as neutrophiles defend the cells from microorganisms, they contain
myeloperoxidase, lysosomal hydrolases. About the relations of eosinophiles with
testifies the growth of their amount during the sensitization of organism, i.e.
during bronchial asthma, helminthiasis. They are able to pile and splits
histamine, “to dissolve” thrombus with the participation of plasminogen and
bradykinin-kininase.
Monocytes
They are formed in the bone marrow. They make up 4-8% of all leukocytes.
According to the function they are called macrophages. Tissue macrophages
derive from blood monocytes. Depending on their position they are called: in
the liver – reticuloendotheliocytes, in the lungs - alveolar macrophages, in
the intermediate substance of connective tissue – histocytes etc. Monocytes are
characterized by a wide set of lysosomal
enzymes with the optimum activity in the acidic condition. The major
functions of monocytes and macrophages are endocytosis and phagocytosis.
Lymphocytes
The amount – 20-25%, are formed in the lymphoid tissue
or thymus, play important role in the formation of humoral and cellular
immunity. Lymphocytes have powerful system of synthesis of antibody proteins,
energy is majorily pertained due to glycolysis, rarely – by aerobic way.
http://www.youtube.com/watch?v=cD_uAGPBfQQ&feature=related
There
are several kinds of lymphocytes (although they all look alike under the
microscope), each with different functions to perform . The most common types
of lymphocytes are
·
B lymphocytes
("B cells"). These are responsible for making antibodies.
·
T lymphocytes
("T cells"). There are several subsets of these:
o
inflammatory T cells
that recruit macrophages and neutrophils to the site of infection or other
tissue damage
o
cytotoxic T lymphocytes
(CTLs) that kill virus-infected and, perhaps, tumor cells
o
helper T cells
that enhance the production of antibodies by B cells
Although bone
marrow is the ultimate source of lymphocytes, the lymphocytes that will become
T cells migrate from the bone marrow to the thymus where they mature. Both B cells and T cells
also take up residence in lymph nodes, the spleen and other tissues where they
·
encounter antigens;
·
continue to divide by mitosis;
·
mature into fully functional cells.
Monocytes
Monocytes
leave the blood and become macrophages and dendritic cells.
This
scanning electron micrograph (courtesy of Drs. Jan M. Orenstein and Emma
Shelton) shows a single macrophage surrounded by several lymphocytes.
Macrophages
are large, phagocytic cells that engulf
·
foreign material (antigens) that
enter the body
·
dead and dying cells of the body.
Thrombocytes
(blood platelets)
Platelets
are cell fragments produced from megakaryocytes.
Blood
normally contains 150,000–350,000 per microliter (µl) or cubic millimeter (mm3).
This number is normally maintained by a homeostatic (negative-feedback)
mechanism .
The amount – less than 1%, they play the main role in
the process of hemostasis. They are formed as a result of disintegration of
megakaryocytes in the bone marrow. Their
–life-time is 7-9 days. In spite of the fact that thrombocytes have no nucleus,
they are able to perform practically all functions of the cell, besides DNA
synthesis.
If
this value should drop much below 50,000/µl, there is a danger of uncontrolled
bleeding because of the essential role that platelets have in blood clotting.
Some
causes:
·
certain drugs and herbal remedies;
·
autoimmunity.
When
blood vessels are cut or damaged, the loss of blood from the system must be
stopped before shock
and possible death occur. This is accomplished by solidification of the blood,
a process called coagulation or clotting.
A blood
clot consists of
·
a plug of platelets enmeshed
in a
·
network of insoluble fibrin
molecules.
Red Blood Cells (erythrocytes)
The
most numerous type in the blood.
·
Women average about 4.8 million of
these cells per cubic millimeter (mm3; which is the same as a
microliter [µl]) of blood.
·
Men average about 5.4 x 106
per µl.
·
These values can vary over quite a
range depending on such factors as health and altitude. (Peruvians living at 18,000 feet may
have as many as 8.3 x 106 RBCs per µl.)
RBC
precursors mature in the bone marrow closely attached to a macrophage.
·
They manufacture hemoglobin until it
accounts for some 90% of the dry weight of the cell.
·
The nucleus is squeezed out of the cell
and is ingested by the macrophage.
·
No-longer-needed proteins are
expelled from the cell in vesicles called exosomes.
Human blood contains 25 trillion of erythrocytes.
Their main function – transportation of O2 and CO2 – they
perform due to the fact that they contain 34% of hemoglobin, and per dry cells
mass – 95%. The total amount of
hemoglobin in the blood equals 130-160 g/l. In the process of erythropoesis the
preceding cells decrease their size. Their nuclei at the end of the process are
ruined and pushed out of the cells. 90% of glucose in the erythrocytes is
decomposed in the process of glycolysis and 10% - by pentose-phosphate way.
There are noted congenital defects of enzymes of these metabolic ways of
erythrocytes. During this are usually observed hemolytic anemia and other
structural and functional erythrocytes’ affections.
This
scanning electron micrograph (courtesy of Dr. Marion J. Barnhart) shows the
characteristic biconcave shape of red blood cells.
Thus RBCs are
terminally differentiated; that is, they can never divide. They live about 120
days and then are ingested by phagocytic cells in the liver and spleen. Most of
the iron in their hemoglobin is reclaimed for reuse. The remainder of the heme
portion of the molecule is degraded into bile pigments
and excreted by the liver. Some 3 million RBCs die and are scavenged by the
liver each second.
Red
blood cells are responsible for the transport of oxygen and carbon
dioxide.
Oxygen Transport
In
adult humans the hemoglobin (Hb) molecule
·
consists of four polypeptides:
o
two alpha (α)
chains of 141 amino acids and
o
two beta (β)
chains of 146 amino acids
·
Each of these
is attached the prosthetic group heme.
·
There is one atom of iron at the center
of each heme.
·
One molecule of oxygen can bind to
each heme.
http://www.youtube.com/watch?v=WXOBJEXxNEo&feature=related
The
reaction is reversible.
·
Under the conditions of
lower temperature, higher pH, and increased oxygen pressure in the capillaries
of the lungs, the reaction proceeds to the right. The purple-red deoxygenated
hemoglobin of the venous blood becomes the bright-red oxyhemoglobin of
the arterial blood.
·
Under the conditions of
higher temperature, lower pH, and lower oxygen pressure in the tissues, the
reverse reaction is promoted and oxyhemoglobin gives up its oxygen.
Carbon Dioxide Transport
Carbon
dioxide (CO2) combines with water forming carbonic acid, which
dissociates into a hydrogen ion (H+) and a bicarbonate ions
:
CO2
+ H2O ↔ H2CO3 ↔ H+ +
HCO3−
95%
of the CO2 generated in the tissues is carried in the red blood
cells:
·
It
probably enters (and leaves) the cell by diffusing through transmembrane
channels in the plasma membrane. (One of the proteins that forms the channel is
the D antigen that is the most important factor in the Rh system
of blood groups.)
·
Once
inside, about one-half of the CO2 is directly bound to hemoglobin
(at a site different from the one that binds oxygen).
·
The
rest is converted — following the equation above — by the enzyme carbonic
anhydrase into
o
bicarbonate
ions that diffuse back out into the plasma and
o
hydrogen
ions (H+) that bind to the protein portion of the hemoglobin (thus
having no effect on pH).
Only
about 5% of the CO2 generated in the tissues dissolves directly in
the plasma. (A good thing, too: if all the CO2 we make were carried
this way, the pH of the blood would drop from its normal 7.4 to an
instantly-fatal 4.5!)
When
the red cells reach the lungs, these reactions are reversed and CO2
is released to the air of the alveoli.
Anemia
is a shortage of
·
RBCs
and/or
·
the
amount of hemoglobin in them.
Anemia
has many causes. One of the most common is an inadequate intake of iron
in the diet.
Red
blood cells have surface antigens that differ between people and that create
the so-called blood groups such as the ABO system and the Rh
system.
An
Essay on Hemoglobin Structure and Function:
Figure 1 is a model of human
deoxyhemoglobin. It was created in RasMol version 2.6 by Roger Sayle
using the pdb coordinates from the pdb file 4hhb. The 3D coordinates were
determed from x-ray crystallography by Fermi, G., Perutz, M. F., Shaanan, B.,
Fourme, R.: The crystal structure of human deoxyhaemoglobin at 1.74 A
resolution. J Mol Biol 175 pp. 159 (1984)
Hemoglobin
is the protein that carries oxygen from the lungs to the tissues and carries
carbon dioxide from the tissues back to the lungs. In order to function most
efficiently, hemoglobin needs to bind to oxygen tightly in the oxygen-rich
atmosphere of the lungs and be able to release oxygen rapidly in the relatively
oxygen-poor environment of the tissues. It does this in a most elegant and
intricately coordinated way. The story of hemoglobin is the prototype example
of the relationship between structure and function of a protein molecule.
Hemoglobin
Structure
A
hemoglobin molecule consists of four polypeptide chains: two alpha chains, each
with 141 amino acids and two beta chains, each with 146 amino acids. The
protein portion of each of these chains is called "globin". The a and b globin chains are very similar in
structure. In this case, a and b refer to the two types of globin. Students
often confuse this with the concept of a helix and b sheet secondary
structures. But, in fact, both the a and b globin chains contain primarily a
helix secondary structure with no b sheets.
Figure 2 is a close up view of one
of the heme groups of the human a chain from dexoyhemoglobin. In this
view, the iron is coordinated by a histidine side chain from amino acid 87
(shown in green.)
Each
a or b globin chain folds into 8 a helical segments (A-H) which, in turn, fold
to form globular tertiary structures that look roughly like sub-microscopic
kidney beans. The folded helices form a pocket that holds the working part of
each chain, the heme.
http://www.youtube.com/watch?v=eor6EK_JP40
A heme group is a flat ring molecule containing
carbon, nitrogen and hydrogen atoms, with a single Fe2+ ion at the
center. Without the iron, the ring is called a porphyrin. In a heme molecule,
the iron is held within the flat plane by four nitrogen ligands from the
porphyrin ring. The iron ion makes a fifth bond to a histidine side chain from
one of the helices that form the heme pocket. This fifth coordination bond is
to histidine 87 in the human a chain and histidine 92 in the human b chain.
Both histidine residues are part of the F helix in each globin chain. t
The
Bohr Effect
The
ability of hemoglobin to release oxygen, is affected by pH, CO2 and
by the differences in the oxygen-rich environment of the lungs and the
oxygen-poor environment of the tissues. The pH in the tissues is considerably
lower (more acidic) than in the lungs. Protons are generated from the reaction
between carbon dioxide and water to form bicarbonate:
CO2
+ H20 -----------------> HCO3- + H+
This
increased acidity serves a twofold purpose. First, protons lower the affinity
of hemoglobin for oxygen, allowing easier release into the tissues. As all four
oxygens are released, hemoglobin binds to two protons. This helps to maintain
equilibrium towards the right side of the equation. This is known as the Bohr
effect, and is vital in the removal of carbon dioxide as waste because CO2
is insoluble in the bloodstream. The bicarbonate ion is much more soluble, and
can thereby be transported back to the lungs after being bound to hemoglobin.
If hemoglobin couldn’t absorb the excess protons, the equilibrium would shift
to the left, and carbon dioxide couldn’t be removed.
In
the lungs, this effect works in the reverse direction. In the presence of the
high oxygen concentration in the lungs, the proton affinity decreases. As
protons are shed, the reaction is driven to the left, and CO2 forms
as an insoluble gas to be expelled from the lungs. The proton poor hemoglobin
now has a greater affinity for oxygen, and the cycle continues.
Haemoglobin or hemoglobin
(frequently abbreviated as Hb or Hgb) is the iron-containing
oxygen-transport
metalloprotein
in the red
blood cells of the blood
in vertebrates
and other animals; in mammals
the protein makes up about 97% of the red cell’s dry content, and around 35% of
the total content including water. Hemoglobin transports oxygen from the lungs
or gills
to the rest of the body, such as to the muscles,
where it releases the oxygen load. Hemoglobin also has a variety of other
gas-transport and effect-modulation duties, which vary from species to species,
and which in invertebrates may be quite diverse.
The
name hemoglobin is the concatenation of heme and globin,
reflecting the fact that each subunit
of hemoglobin is a globular
protein with an embedded heme
(or haem) group; each heme group contains an iron
atom, and this is responsible for the binding of oxygen. The most common type
of hemoglobin in mammals contains four such subunits, each with one heme group.
Mutations
in the genes
for the hemoglobin protein in humans result in a group of hereditary
diseases termed the hemoglobinopathies,
the most common members of which are sickle-cell
disease and thalassemia.
Historically in human medicine, the hemoglobinopathy of sickle-cell
disease was the first disease to be understood in its mechanism of
dysfunction, completely down to the molecular level. However, not all of such
mutations produce disease states, and are formally recognized as hemoglobin
variants (not diseases).[]
Hemoglobin
(Hb) is synthesized in a complex series of steps. The heme
portion is sythesized in both the the mitochondria
and cytosol
of the immature red blood cell, while the globin
protein portions of the molecule are sythesized by ribosomes in the cytosol [3].
Production of Hb continues in the cell throughout its early development from
the proerythroblast
to the reticulocyte
in the bone
marrow. At this point, the nucleus is lost in mammals, but not in birds
and many other species. Even after the loss of the nucleus in mammals, however,
residual ribosomal RNA allows further synthesis of Hb until the reticulocyte
loses its RNA soon after entering the vasculature (this hemoglobin-synthetic
RNA in fact gives the reticulocyte
its reticulated appearance and name).
The
empirical chemical formula of the most common human hemoglobin is C2952H4664N812O832S8Fe4,
but as noted above, hemoglobins vary widely across species, and even (through
common mutations) slightly among subgroups of humans.
In
humans, the hemoglobin molecule
is an assembly of four globular
protein subunits. Each subunit
is composed of a protein
chain tightly associated with a non-protein heme
group. Each protein chain arranges into a set of alpha-helix
structural segments connected together in a globin
fold arrangement, so called because this arrangement is the same folding
motif used in other heme/globin proteins such as myoglobin.[4][5]
This folding pattern contains a pocket which strongly binds the heme group.
A
heme group consists of an iron
(Fe) atom held in a heterocyclic
ring, known as a porphyrin.
The iron atom, which is the site of oxygen binding, bonds with the four nitrogens
in the center of the ring, which all lie in one plane. The iron is also bound
strongly to the globular protein via the imidazole
ring of a histidine
residue below the porphyrin ring. A sixth position can reversibly bind oxygen,
completing the octahedral group of six ligands. Oxygen binds in an "end-on
bent" geometry where one oxygen atom binds Fe and the other protrudes at
an angle. When oxygen is not bound, a very weakly bonded water molecule fills
the site, forming a distorted octahedron.
The
iron atom may either be in the Fe2+ or Fe3+ state, but
ferrihemoglobin (methemoglobin)
(Fe3+) cannot bind oxygen. In binding, oxygen temporarily oxidizes
Fe to (Fe3+), so iron must exist in the +2 oxidation state in order
to bind oxygen. The body reactivates hemoglobin found in the inactive (Fe3+)
state by reducing the iron center.
In
adult humans, the most common hemoglobin type is a tetramer
(which contains 4 subunit proteins) called hemoglobin A, consisting of
two α and two β subunits non-covalently bound, each made of 141 and
146 amino acid residues, respectively. This is denoted as α2β2.
The subunits are structurally similar and about the same size. Each subunit has
a molecular weight of about 17,000 daltons,
for a total molecular
weight of the tetramer of about 68,000 daltons. Hemoglobin A is the
most intensively studied of the hemoglobin molecules.
The
four polypeptide
chains are bound to each other by salt
bridges, hydrogen
bonds, and hydrophobic
interactions. There are two kinds of contacts between the α and
β chains: α1β1 and α1β2.
Oxyhemoglobin is formed during respiration when oxygen
binds to the heme
component of the protein hemoglobin in red blood cells. This process occurs in
the pulmonary capillaries adjacent to the alveoli
of the lungs.
The oxygen then travels through the blood stream to be dropped off at cells
where it is utilized in aerobic glycolysis
and in the production of ATP
by the process of oxidative
phosphorylation. It doesn't however help to counteract a decrease in
blood pH. Ventilation,
or breathing, may reverse this condition by removal of carbon dioxide, thus
causing a shift up in pH.[6]
Deoxyhemoglobin is the form of hemoglobin without the bound oxygen.
The absorption
spectra of oxyhemoglobin and deoxyhemoglobin differ. The oxyhemoglobine
has significantly lower absorption of the 660 nm wavelength
than deoxyhemoglobin, while at 940 nm its absorption is slightly higher.
This difference is used for measurement of the amount of oxygen in patient's blood
by an instrument called pulse
oximeter.
The
oxidation state of iron in hemoglobin is always +2. It does not change when
oxygen binds to the deoxy- form.
Assigning
oxygenated hemoglobin's oxidation state is difficult because oxyhemoglobin is
diamagnetic (no net unpaired electrons), but the low-energy electron
configurations in both oxygen and iron are paramagnetic. Triplet oxygen, the
lowest energy oxygen species, has two unpaired electrons in antibonding π*
molecular orbitals. Iron(II) tends to be in a high-spin configuration where
unpaired electrons exist in eg antibonding orbitals. Iron(III) has an
odd number of electrons and necessarily has unpaired electrons. All of these
molecules are paramagnetic (have unpaired electrons), not diamagnetic, so an
unintuitive distribution of electrons must exist to induce diamagnetism.
The
three logical possibilities are:
1)
Low-spin Fe2+ binds to high-energy singlet oxygen. Both low-spin
iron and singlet oxygen are diamagnetic.
2)
High-spin Fe3+ binds to .O2- (the superoxide
ion) and antiferromagnetism oppositely aligns the two unpaired electrons,
giving diamagnetic properties.
3)
Low-spin Fe4+ binds to O22-. Both are
diamagnetic.
X-ray
photoelectron spectroscopy suggests that iron has an oxidation state of
approximately 3.2 and infrared
stretching frequencies of the O-O bond suggests a bond length fitting
with superoxide. The correct oxidation state of iron is thus the +3 state with
oxygen in the -1 state. The diamagnetism in this configuration arises from the
unpaired electron on superoxide aligning antiferromagnetically in the opposite
direction from the unpaired electron on iron. The second choice being correct
is not surprising because singlet oxygen and large separations of charge are
both unfavorably high-energy states. Iron's shift to a higher oxidation state
decreases the atom's size and allows it into the plane of the porphyrin ring,
pulling on the coordinated histidine residue and initiating the allosteric
changes seen in the globulins. The assignment of oxidation state, however, is
only a formalism so all three models may contribute to some small degree.
Early
postulates by bioinorganic chemists claimed that possibility (1) (above) was
correct and that iron should exist in oxidation state II (indeed iron oxidation
state III as methemoglobin, when not accompanied by superoxide .O2-
to "hold" the oxidation electron, is incapable of binding O2).
The iron chemistry in this model was elegant, but the presence of singlet
oxygen was never explained. It was argued that the binding of an oxygen
molecule placed high-spin iron(II) in an octahedral field of strong-field
ligands; this change in field would increase the crystal
field splitting energy, causing iron's electrons to pair into the
diamagnetic low-spin configuration.
Binding
and release of ligands induces a conformational (structural) change in
hemoglobin. Here, the binding and release of oxygen illustrates the structural
differences between oxy- and deoxyhemoglobin, respectively. Only one of the
four heme groups is shown.
As
discussed above, when oxygen binds to the iron center it causes contraction of
the iron atom, and causes it to move back into the center of the porphyrin ring
plane (see moving diagram). At the same time, the porphyrin ring plane itself
is pushed away from the oxygen and toward the imidizole side chain of the
histidine residue interacting at the other pole of the iron. The interaction
here forces the ring plane sideways toward the outside of the tetramer, and also
induces a strain on the protein helix containing the histidine, as it moves
nearer the iron. This causes a tug on this peptide strand which tends to open
up heme units in the remainder of the molecule, so that there is more room for
oxygen to bind at their heme sites.
In
the tetrameric form of normal adult hemoglobin, the binding of oxygen is thus a
cooperative
process. The binding affinity of hemoglobin for oxygen is increased by the
oxygen saturation of the molecule, with the first oxygens bound influencing the
shape of the binding sites for the next oxygens, in a way favorable for binding.
This positive cooperative binding is achieved through steric
conformational changes of the hemoglobin protein complex as discussed above, i.e.
when one subunit protein in hemoglobin becomes oxygenated, this induces a
conformational or structural change in the whole complex, causing the other
subunits to gain an increased affinity for oxygen. As a consequence, the oxygen
binding curve of hemoglobin is sigmoidal,
or S-shaped, as opposed to the normal hyperbolic
curve associated with noncooperative binding.
Hemoglobin's
oxygen-binding capacity is decreased in the presence of carbon
monoxide because both gases compete for the same binding sites on
hemoglobin, carbon monoxide binding preferentially in place of oxygen. Carbon dioxide
occupies a different binding site on the hemoglobin. Through the enzyme carbonic
anhydrase, carbon dioxide reacts with water to give carbonic
acid, which decomposes into bicarbonate
and protons:
CO2
+ H2O → H2CO3 → HCO3-
+ H+
The
sigmoidal shape of hemoglobin's oxygen-dissociation curve results from cooperative
binding of oxygen to hemoglobin.
Hence
blood with high carbon dioxide levels is also lower in pH
(more acidic).
Hemoglobin can bind protons
and carbon dioxide which causes a conformational change in the protein and
facilitates the release of oxygen. Protons bind at various places along the
protein, and carbon dioxide binds at the alpha-amino
group forming carbamate.
Conversely, when the carbon dioxide levels in the blood decrease (i.e., in the
lung capillaries), carbon dioxide and protons are released from hemoglobin,
increasing the oxygen affinity of the protein. This control of hemoglobin's
affinity for oxygen by the binding and release of carbon dioxide and acid, is
known as the Bohr
effect.
The
binding of oxygen is affected by molecules such as carbon
monoxide (CO) (for example from tobacco
smoking, cars and furnaces). CO competes with oxygen at the heme binding
site. Hemoglobin binding affinity for CO is 200 times greater than its affinity
for oxygen, meaning that small amounts of CO dramatically reduces hemoglobin's
ability to transport oxygen. When hemoglobin combines with CO, it forms a very
bright red compound called carboxyhemoglobin.
When inspired air contains CO levels as low as 0.02%, headache and nausea
occur; if the CO concentration is increased to 0.1%, unconsciousness will
follow. In heavy smokers, up to 20% of the oxygen-active sites can be blocked
by CO.
In
similar fashion, hemoglobin also has competitive binding affinity for cyanide
(CN-), sulfur
monoxide (SO), nitrogen
dioxide (NO2), and sulfide (S2-), including hydrogen
sulfide (H2S). All of these bind to iron in heme without
changing its oxidation state, but they nevertheless inhibit oxygen-binding,
causing grave toxicity.
The
iron atom in the heme group must be in the Fe2+ oxidation state to
support oxygen and other gases' binding and transport. Oxidation to Fe3+
state converts hemoglobin into hemiglobin or methemoglobin
(pronounced "MET-hemoglobin"), which cannot bind oxygen. Hemoglobin
in normal red blood cells is protected by a reduction system to keep this from
happening. Nitrogen dioxide and nitrous
oxide are capable of converting a small fraction of hemoglobin to
methemoglobin, however this is not usually of medical importance (nitrogen
dioxide is poisonous by other mechanisms, and nitrous oxide is routinely used
in surgical anesthesia in most people without undue methemoglobin buildup).
In
people acclimated to high altitudes, the concentration of 2,3-bisphosphoglycerate
(2,3-BPG) in the blood is increased, which allows these individuals to deliver
a larger amount of oxygen to tissues under conditions of lower oxygen tension.
This phenomenon, where molecule Y affects the binding of molecule X to a
transport molecule Z, is called a heterotropic allosteric
effect.
A
variant hemoglobin, called fetal
hemoglobin (HbF, α2γ2), is found in the
developing fetus,
and binds oxygen with greater affinity than adult hemoglobin. This means that
the oxygen binding curve for fetal hemoglobin is left-shifted (i.e., a higher
percentage of hemoglobin has oxygen bound to it at lower oxygen tension), in
comparison to that of adult hemoglobin. As a result, fetal blood in the placenta
is able to take oxygen from maternal blood.
Hemoglobin
also carries nitric
oxide in the globin part of the molecule. This improves oxygen delivery
in the periphery and contributes to the control of respiration. NO binds
reversibly to a specific cystein residue in globin; the binding depends on the
state (R or T) of the hemoglobin. The resulting S-nitrosylated hemoglobin
influences various NO-related activities such as the control of vascular
resistance, blood pressure and respiration. NO is released not in the cytoplasm
of erythrocytes but is transported by an anion exchanger called AE1
out of them.[]
When
red
cells reach the end of their life due to aging or defects, they are
broken down, the hemoglobin molecule is broken up and the iron gets recycled.
When the porphyrin ring is broken up, the fragments are normally secreted in
the bile
by the liver.
This process also produces one molecule of carbon
monoxide for every molecule of heme degraded [4]; this is one of the few
natural sources of carbon
monoxide production in the human body, and is responsible for the normal
blood levels of carbon monoxide even in people breathing pure air. The other
major final product of heme degradation is bilirubin.
Increased levels of this chemical are detected in the blood if red cells are
being destroyed more rapidly than usual. Improperly degraded hemoglobin protein
or hemoglobin that has been released from the blood cells too rapidly can clog
small blood vessels, especially the delicate blood filtering vessels of the kidneys,
causing kidney damage
Decrease
of hemoglobin, with or without an absolute decrease of red
blood cells, leads to symptoms of anemia.
Anemia has many different causes, although iron
deficiency and its resultant iron
deficiency anemia are the most common causes in the Western world. As
absence of iron decreases heme
synthesis, red blood cells in iron deficiency anemia are hypochromic (lacking
the red hemoglobin pigment) and microcytic (smaller than normal). Other anemias
are rarer. In hemolysis
(accelerated breakdown of red blood cells), associated jaundice
is caused by the hemoglobin metabolite bilirubin,
and the circulating hemoglobin can cause renal
failure.
Some
mutations in the globin chain are associated with the hemoglobinopathies,
such as sickle-cell
disease and thalassemia.
Other mutations, as discussed at the beginning of the article, are benign and
are referred to merely as hemoglobin
variants.
There
is a group of genetic disorders, known as the porphyrias
that are characterized by errors in metabolic pathways of heme synthesis. King George
III of the United Kingdom was probably the most famous porphyria
sufferer.
To
a small extent, hemoglobin A slowly combines with glucose
at a certain location in the molecule. The resulting molecule is often referred
to as Hb
A1c. As the concentration
of glucose in the blood increases, the percentage of Hb A that turns into Hb
A1c increases. In diabetics
whose glucose usually runs high, the percent Hb A1c also runs high. Because of
the slow rate of Hb A combination with glucose, the Hb A1c percentage is
representative of glucose level in the blood averaged over a longer time (the
half-life of red blood cells, which is typically 50-55 days).
Hemoglobin
levels are amongst the most commonly performed blood
tests, usually as part of a full blood count or complete
blood count. Results are reported in g/L,
g/dL
or mol/L.
For conversion, 1 g/dL is 0.621 mmol/L. If the total hemoglobin concentration
in the blood falls below a set point, this is called anemia.
Normal values for hemoglobin levels are:
·
Women:
12.1 to 15.1 g/dl
·
Men:
13.8 to 17.2 g/dl
·
Children:
11 to 16 g/dl
·
Pregnant
women: 11 to 12 g/dl
Anemias
are further subclassified by the size of the red blood cells, which are the
cells which contain hemoglobin in vertebrates. They can be classified as
microcytic (small sized red blood cells), normocytic (normal sized red blood
cells), or macrocytic (large sized red blood cells). The hemaglobin is the
typical test used for blood
donation. A comparison with the hematocrit
can be made by multiplying the hemaglobin by three. For example, if the
hemaglobin is measured at 17, that compares with a hematocrit of .51
Glucose
levels in blood can vary widely each hour, so one or only a few samples from a
patient analyzed for glucose may not be representative of glucose control in
the long run. For this reason a blood sample may be analyzed for Hb A1c
level, which is more representative of glucose control averaged over a longer
time period (determined by the half-life of the individual's red blood cells,
which is typically 50-55 days). People whose Hb A1c runs 6.0% or
less show good longer-term glucose control. Hb A1c values which are
more than 7.0% are elevated. This test is especially useful for diabetics.[8]
BIOLOGICAL
BUFFERS OF BLOOD
Acid–base
homeostasis is the part of human homeostasis concerning the proper balance
between acids and bases, also called body pH. The body is very sensitive to its
pH level, so strong mechanisms exist to maintain it. Outside the acceptable
range of pH, proteins are denatured and digested, enzymes lose their ability to
function, and death may occur.
A
buffer solution is an aqueous solution consisting of a mixture of a weak acid
and its conjugate base or a weak base and its conjugate acid. Its pH changes
very little when a small amount of strong acid or base is added to it. Buffer
solutions are used as a means of keeping pH at a nearly constant value in a
wide variety of chemical applications.
Many
life forms thrive only in a relatively small pH range so they utilize a buffer
solution to maintain a constant pH. One example of a buffer solution found in
nature is blood. The body's acid–base balance is normally tightly regulated,
keeping the arterial blood pH between 7.38 and 7.42. Several buffering agents
that reversibly bind hydrogen ions and impede any change in pH exist.
Extracellular buffers include bicarbonate and ammonia, whereas proteins and
phosphate act as intracellular buffers. The bicarbonate buffering system is
especially key, as carbon dioxide (CO2) can be shifted through
carbonic acid (H2CO3) to hydrogen ions and bicarbonate
(HCO3-):
Acid–base
imbalances that overcome the buffer system can be compensated in the short term
by changing the rate of ventilation. This alters the concentration of carbon
dioxide in the blood, shifting the above reaction according to Le Chatelier's
principle, which in turn alters the pH.
The
kidneys are slower to compensate, but renal physiology has several powerful
mechanisms to control pH by the excretion of excess acid or base. In response
to acidosis, tubular cells reabsorb more bicarbonate from the tubular fluid,
collecting duct cells secrete more hydrogen and generate more bicarbonate, and
ammoniagenesis leads to increased formation of the NH3 buffer. In responses to alkalosis, the
kidneys may excrete more bicarbonate by decreasing hydrogen ion secretion from
the tubular epithelial cells, and lowering rates of glutamine metabolism and
ammonium excretion.
This
huge buffer capacity has another not immediately obvious implication for how we
think about the severity of an acid-base disorder. You would think that the
magnitude of an acid-base disturbance could be quantified merely by looking at
the change in [H+] - BUT this is not so.
Because
of the large buffering capacity, the actual change in [H+] is so
small it can be ignored in any quantitative assessment, and instead, the magnitude
of a disorder has to be estimated indirectly from the decrease in the total
concentration of the anions involved in the buffering. The buffer anions,
represented as A-, decrease because they combine stoichiometrically
with H+ to produce HA.
A decrease in A- by 1
mmol/l represents a 1,000,000 nano-mol/l amount of H+ that is hidden from view and this is
several orders of magnitude higher than the visible few nanomoles/l change in
[H+] that is visible.) - As noted above in the comments about the
Swan & Pitts experiment, 13,999,994 out of 14,000,000 nano-moles/l of H+ were hidden on buffers and just to
count the 36 that were on view would give a false impression of the magnitude
of the disorder.
The
major buffer system in the ECF is the CO2-bicarbonate buffer system.
This is responsible for about 80% of extracellular buffering. It is the most
important ECF buffer for metabolic acids but it cannot buffer respiratory
acid-base disorders.
The
components are easily measured and are related to each other by the
Henderson-Hasselbalch equation.
Henderson-Hasselbalch
Equation
pH = pK’a + log10 (
[HCO3] / 0.03 x pCO2)
The
pK’a value is dependent on the temperature, [H+] and the ionic
concentration of the solution. It has a value of 6.099 at a temperature of 37C
and a plasma pH of 7.4. At a temperature of 30C and pH of 7.0, it has a value
of 6.148. For practical purposes, a value of 6.1 is generally assumed and
corrections for temperature, pH of plasma and ionic strength are not used
except in precise experimental work.
The
pK'a is derived from the Ka value of the following reaction:
CO2 + H2O <=> H2CO3 <=> H+ + HCO3-
(where
CO2 refers to
dissolved CO2)
The
concentration of carbonic acid is very low compared to the other components so
the above equation is usually simplified to:
CO2 + H2O <=> H+ + HCO3-
By
the Law of Mass Action:
Ka
= [H+] . [HCO3-] / [CO2] . [H20]
The
concentration of H2O is so large (55.5M) compared to the other
components, the small loss of water due to this reaction changes its
concentration by only an extremely small amount. This means that [H2O]
is effectively constant. This allows further simplification as the two
constants (Ka and [H2O] ) can be combined into a new constant K’a.
K’a
= Ka x [H2O] = [H+] . [HCO3-] / [CO2]
Substituting:
K'a
= 800 nmol/l (value for plasma at 37C)
[CO2]
= 0.03 x pCO2 (by
Henry’s Law) [where 0.03 is the solubility coefficient]
into
the equation yields the Henderson Equation:
[H+]
= (800 x 0.03) x pCO2 /
[HCO3-] = 24 x pCO2 /
[HCO3-] nmol/l
Taking
the logs (to base 10) of both sides yields the Henderson-Hasselbalch equation:
pH
= log10(800) - log (0.03 pCO2 /
[HCO3-] )
pH
= 6.1 + log ( [HCO3] / 0.03 pCO2 )
On
chemical grounds, a substance with a pKa of 6.1 should not be a good buffer at
a pH of 7.4 if it were a simple buffer. The system is more complex as it is
‘open at both ends’ (meaning both [HCO3] and pCO2 can be adjusted) and this greatly
increases the buffering effectiveness of this system. The excretion of CO2 via the lungs is particularly
important because of the rapidity of the response. The adjustment of pCO2 by
change in alveolar ventilation has been referred to as physiological buffering.
The
other buffer systems in the blood are the protein and phosphate buffer systems.
These
are the only blood buffer systems capable of buffering respiratory acid-base
disturbances as the bicarbonate system is ineffective in buffering changes in H+ produced by itself.
The
concentration of phosphate in the blood is so low that it is quantitatively
unimportant. Phosphates are important buffers intracellularly and in urine
where their concentration is higher.
Phosphoric
acid is triprotic weak acid and has a pKa value for each of the three
dissociations:
The
three pKa values are sufficiently different so that at any one pH only the
members of a single conjugate pair are present in significant concentrations.
At
the prevailing pH values in most biological systems, monohydrogen phosphate
(HPO4-2) and dihydrogen phosphate (H2PO4-)
are the two species present. The pKa2 is 6.8 and this makes the closed
phosphate buffer system a good buffer intracellularly and in urine. The pH of
glomerular ultrafiltrate is 7.4 and this means that phosphate will initially be
predominantly in the monohydrogen form and so can combine with more H+ in the renal tubules. This makes the
phosphate buffer more effective in buffering against a drop in pH than a rise
in pH.
Note:
The ‘true’ pKa2 value is actually 7.2 if measured at zero ionic strength but at
the typical ionic strength found in the body its apparent value is 6.8. The
other factor which makes phosphate a more effective buffer intracellularly and
in urine is that its concentration is much higher here than in extracellular
fluid.
Protein
buffers in blood include haemoglobin (150g/l) and plasma proteins (70g/l).
Buffering is by the imidazole group of the histidine residues which has a pKa
of about 6.8. This is suitable for effective buffering at physiological pH.
Haemoglobin is quantitatively about 6 times more important then the plasma
proteins as it is present in about twice the concentration and contains about
three times the number of histidine residues per molecule. For example if blood
pH changed from 7.5 to 6.5, haemoglobin would buffer 27.5 mmol/l of H+ and total plasma protein buffering
would account for only 4.2 mmol/l of H+.
Deoxyhaemoglobin
is a more effective buffer than oxyhaemoglobin and this change in buffer
capacity contributes about 30% of the Haldane effect. The major factor
accounting for the Haldane effect in CO2 transport is the much greater ability
of deoxyhaemoglobin to form carbamino compounds.
This
buffer functions in exactly the same way as the phosphate buffer. Additional H+ is consumed by HCO3- and additional OH- is consumed by H2CO3.
The value of Ka for this equilibrium is 7.9 Ч 10-7,
and the pKa is
6.1 at body temperature. In blood plasma, the concentration of hydrogen
carbonate ion is about twenty times the concentration of carbonic acid. The pH
of arterial blood plasma is 7.40. If the pH falls below this normal value, a
condition called acidosis is produced. If the pH rises above the
normal value, the condition is called alkalosis.
The
concentrations of hydrogen carbonate ions and of carbonic acid are controlled
by two independent physiological systems. Carbonic acid concentration is
controlled by respiration, that is through the lungs. Carbonic acid is in
equilibrium with dissolved carbon dioxide gas.
H2CO3(aq) CO2(aq) + H2O(l)
An
enzyme called carbonic anhydrase catalyzes the conversion of carbonic acid to
dissolved carbon dioxide. In the lungs, excess dissolved carbon dioxide is
exhaled as carbon dioxide gas.
CO2(aq) CO2(g)
The
concentration of hydrogen carbonate ions is controlled through the kidneys. Excess
hydrogen carbonate ions are excreted in the urine.
The
much higher concentration of hydrogen carbonate ion over that of carbonic acid
in blood plasma allows the buffer to respond effectively to the most common
materials that are released into the blood. Normal metabolism releases mainly
acidic materials: carboxylic acids such as lactic acid (HLac). These acids
react with hydrogen carbonate ion and form carbonic acid.
HLac(aq)
+ HCO3-(aq) Lac-(aq) + H2CO3(aq)
The
carbonic acid is converted through the action of the enzyme carbonic anhydrase
into aqueous carbon dioxide.
H2CO3(aq) CO2(aq) + H2O(l)
An
increase in CO2(aq) concentration stimulates increased breathing,
and the excess carbon dioxide is released into the air in the lungs.
The
condition called respiratory
acidosis occurs when blood
pH falls as a result of decreased respiration. When respiration is restricted,
the concentration of dissolved carbon dioxide in the blood increases, making
the blood too acidic. Such a condition can be produced by asthma, pneumonia,
emphysema, or inhaling smoke.
Metabolic
acidosis is the decrease in
blood pH that results when excessive amounts of acidic substances are released
into the blood. This can happen through prolonged physical exertion, by
diabetes, or restricted food intake. The normal body response to this condition
is increases breathing to reduce the amount of dissolved carbon dioxide in the
blood. This is why we breathe more heavily after climbing several flights of
stairs.
Respiratory
alkalosis results from
excessive breathing that produces an increase in blood pH. Hyperventilation
causes too much dissolved carbon dioxide to be removed from the blood, which
decreases the carbonic acid concentration, which raises the blood pH. Often,
the body of a hyperventilating person will react by fainting, which slows the
breathing.
Metabolic
alkalosis is an increase in
blood pH resulting from the release of alkaline materials into the blood. This
can result from the ingestion of alkaline materials, and through overuse of
diuretics. Again, the body usually responds to this condition by slowing
breathing, possibly through fainting.
The
carbonic acid-hydrogen carbonate ion buffer works throughout the body to
maintain the pH of blood plasma close to 7.40. The body maintains the buffer by
eliminating either the acid (carbonic acid) or the base (hydrogen carbonate
ions). Changes in carbonic acid concentration can be effected within seconds
through increased or decreased respiration. Changes in hydrogen carbonate ion
concentration, however, require hours through the relatively slow elimination
through the kidneys
Plasma is the straw-colored liquid in which the blood
cells are suspended.
Plasma
transports materials needed by cells and materials that must be removed from
cells:
·
various
ions (Na+, Ca2+, HCO3−, etc.
·
glucose
and traces of other sugars
·
amino
acids
·
other
organic acids
·
cholesterol
and other lipids
·
hormones
·
urea
and other wastes
Most
of these materials are in transit from a place where they are added to the
blood (a "source")
·
exchange
organs like the intestine
·
depots
of materials like the liver
to
places ("sinks") where they will be removed from the blood.
·
every
cell
·
exchange organs like the kidney, and
skin.
Proteins
make up 6–8% of the blood. They are about equally divided between serum
albumin and a great variety of serum globulins.
After
blood is withdrawn from a vein and allowed to clot, the clot slowly shrinks. As
it does so, a clear fluid called serum is squeezed out. Thus:
Serum is blood plasma without fibrinogen and other
clotting factors.
The
serum proteins can be separated by electrophoresis.
·
A
drop of serum is applied in a band to a thin sheet of supporting material, like
paper, that has been soaked in a slightly-alkaline salt solution.
· At
pH 8.6, which is commonly used, all the proteins are negatively charged, but
some more strongly than others.
·
A
direct current can flow through the paper because of the conductivity of the
buffer with which it is moistened.
·
As
the current flows, the serum proteins move toward the positive electrode.
·
The
stronger the negative charge on a protein, the faster it migrates.
·
After
a time (typically 20 min), the current is turned off and the proteins stained
to make them visible (most are otherwise colorless).
·
The
separated proteins appear as distinct bands.
·
The
most prominent of these and the one that moves closest to the positive
electrode is serum albumin.
·
Serum
albumin
o
is
made in the liver
o
binds
many small molecules for transport through the blood
o
helps
maintain the osmotic pressure
of the blood
·
The
other proteins are the various serum globulins.
·
They
migrate in the order
o
alpha
globulins (e.g., the proteins that transport thyroxine and retinol [vitamin A])
o
beta
globulins (e.g., the iron-transporting protein
transferrin)
o
gamma
globulins.
§
Gamma
globulins are the least negatively-charged serum proteins. (They are so weakly
charged, in fact, that some are swept in the flow of buffer back toward the
negative electrode.)
§
Most
antibodies are gamma globulins.
§
Therefore
gamma globulins become more abundant following infections or immunizations.
Albumins – multidispersed fraction of blood plasma which are characterized by the high electrophoretic mobility and
mild dissolubility in water and saline solutions. Molecular weight of albumins
is about 60000. Due to high hydrophilic properties albumins bind a significant amount of
water, and the volume of their molecule under hydratation is doubled. Hydrative layer formed around
the serum albumins provides to 70-80 % of oncotic pressure of blood plasma
proteins, that can be applied in clinical practice at albumins transfusion to
patients with tissue edemas. The decreasing of albumins concentration in blood
plasma, for example under disturbance of their synthesis in hepatocytes at liver failure, can cause the water transition from a vessels
into the tissues and development of oncotic edemas.
Albumins execute also important physiological
function as transporters of a lot of metabolites and diverse low molecular
weight structures. The molecules of albumins have several sites with centers of linkage for
molecules of organic ligands, which are affixed by the electrostatic and hydrophobic bonds. Serum albumins can affix and convey fatty acids, cholesterol,
cholic pigments (bilirubin and that similar), vitamins, hormones, some amino acids, toxins and medicines.
Albumins also execute the buffer function. Due to
the availability in their structure amino and carboxylic groups albumins can
react both as acids and as alkaline.
Albumins can bound different toxins in blood
plasma (bilirubin, foreign substances et c.). This is the desintoxicative function of albumins.
Albumins also play role of amino acids depot in
the organism. They can supply amino acids for the building of another proteins,
for example enzymes.
Globulins -
heterogeneous fraction of blood proteins which execute transport (a1-globulins – transport of lipids, thyroxin, corticosteroid hormones; a2-globulins - transport of lipids, copper ions; b-globulins - transport of lipids, iron) and
protective (participation of b-globulins in immune reactions as antitoxins; g-globulins as
immunoglobulins) functions. They also support the blood oncotic pressure and acid-alkaline
balance, provide amino acids for the organism requirements. The molecular
weight of globulins is approximately 150000-300000.
The globulin level in blood plasma is 20-40 g/l. A
ratio between concentrations of albumins and globulins (so called “protein
coefficient”) in blood plasma is often determined in clinical practice. In
healthy people this coefficient is 1,5-2,0.
Fibrinogen – important protein of blood plasma,
precursor of fibrin, the structural element of blood clots. Fibrinogen
participates in blood clotting and thus prevents the loss of blood from the
vascular system of vertebrates. The approximate molecular weight of fibrinogen
is 340000. It is the complex protein, it contains the carbohydrate as
prosthetic group. The content of firinogen in blood is 3-4 g/l.
Subfractions
of a1, a2, b
and g globulins,
their structure and functions.
Immunoglobulins (Ig A, Ig G, Ig E, Ig M) - proteins of g-globulin
fraction of blood plasma executing the functions of antibodies which are the main
effectors of humoral immunity. They appear in the blood serum and certain cells
of a vertebrate in response to the introduction of a protein or some other
macromolecule foreign to that species.
Immunoglobulin molecules have bindind sites that
are specific for and complementary to the structural features of the antigen
that induced their formation. Antibodies are highly specific for the foreign
proteins that evoke their formation.
Molecules of immunoglobulins are glycoproteins. The protein part of
immunoglobulins contain four polipeptide
chains: two heavy H-chains and two light L-chains.
The acute phase response develops in a wide
range of acute and chronic inflammatory conditions like bacterial, viral, or
fungal infections; rheumatic and other inflammatory diseases; malignancy; and
tissue injury or necrosis. These conditions cause release of interleukin-6 and
other cytokines that trigger the synthesis of CRP and fibrinogen by the liver.
During the acute phase response, levels of CRP rapidly increase within 2 hours
of acute insult, reaching a peak at 48 hours. With resolution of the acute phase
response, CRP declines with a relatively short half-life of 18 hours. Measuring
CRP level is a screen for infectious and inflammatory diseases. Rapid, marked
increases in CRP occur with inflammation, infection, trauma and tissue
necrosis, malignancies, and autoimmune disorders. Because there are a large
number of disparate conditions that can increase CRP production, an elevated
CRP level does not diagnose a specific disease. An elevated CRP level can
provide support for the presence of an inflammatory disease, such as rheumatoid
arthritis, polymyalgia rheumatica or giant-cell
arteritis.
The physiological
role of CRP is to bind to phosphocholine expressed on the surface of dead or
dying cells (and some types of bacteria) in order to activate the complement
system. CRP binds to phosphocholine on microbes and damaged cells and enhances
phagocytosis by macrophages. Thus, CRP participates in the clearance of
necrotic and apoptotic cells.
CRP is a member of
the class of acute-phase reactants, as its levels rise dramatically during inflammatory processes occurring in the body. This
increment is due to a rise in the plasma concentration of IL-6,
which is produced predominantly by macrophages[2] as well asadipocytes. CRP binds to phosphocholine on microbes. It is thought to assist
in complement binding to foreign and damaged cells
and enhances phagocytosis by macrophages (opsonin
mediated phagocytosis), which express a receptor for CRP. It is also
believed to play another important role in innate
immunity, as an early defense system against infections.
CRP rises up to
50,000-fold in acute inflammation, such as infection. It rises above normal
limits within 6 hours, and peaks at 48 hours. Its half-life is constant, and
therefore its level is mainly determined by the rate of production (and hence
the severity of the precipitating cause).
Serum
amyloid A is a related
acute-phase marker that responds rapidly in similar circumstances.
C-reactive protein (g-fraction).
This protein received the title owing to its capacity to react with
C-polysaccharide of a pneumococcus forming precipitates. According to its
chemical nature C-reactive protein is glycoprotein.
C-reactive
protein, pentraxin-related
CRP is used mainly
as a marker of inflammation. Apart from liver
failure, there are few known factors that interfere with CRP production.[2]
Measuring and
charting CRP values can prove useful in determining disease progress or the
effectiveness of treatments. Blood,
usually collected in a serum-separating
tube, is analysed in amedical
laboratory or at the point
of care. Various analytical methods are available for CRP determination, such
as ELISA, immunoturbidimetry,
rapid immunodiffusion,
and visual agglutination.
Reference
ranges for blood tests, showing C-reactive protein in brown-yellow in
center.
A high-sensitivity
CRP (hs-CRP) test measures low levels of CRP using laser nephelometry.
The test gives results in 25 minutes with a sensitivity down to 0.04 mg/L.
Normal concentration
in healthy human serum is usually lower than 10 mg/L, slightly increasing
with aging.
Higher levels are found in late pregnant women, mild inflammation and viral
infections (10–40 mg/L),
active inflammation, bacterial infection (40–200 mg/L), severe bacterial
infections and burns (>200 mg/L).[26]
CRP is a more
sensitive and accurate reflection of the acute phase response than the ESR (Erythrocyte
Sedimentation Rate). The half-life of CRP is constant. Therefore, CRP
level is mainly determined by the rate of production (and hence the severity of
the precipitating cause). In the first 24 h, ESR may be normal and CRP elevated.
CRP returns to normal more quickly than ESR in response to therapy.
Arterial damage
results from white
blood cell invasion and inflammation within the wall. CRP is a general
marker for inflammation and infection, so it can be used as a very rough proxy
for heart disease risk. Since many things can cause elevated CRP, this is not a
very specific prognostic indicator.[27] Nevertheless, a level above
2.4 mg/L has been associated with a doubled risk of a coronary event
compared to levels below 1 mg/L;[2] however, the study group in this case
consisted of patients who had been diagnosed with unstable angina pectoris;
whether elevated CRP has any predictive value of acute coronary events in the
general population of all age ranges remains unclear.
Crioglobulin - the protein of the g-globulin fraction. Like to the C-reactive protein
crioglobulin absent in blood plasma of the healthy people and occurs at
leukoses, rheumatic disease, liver cirrhosis, nephroses. The characteristic
physico-chemical feature of crioglobulin is its dissolubility at standard body
temperature (37 oC) and capacity to form the sediment at cooling of
a blood plasma up to 4 oC.
a2-macroglobulin - protein of a2-globulin fraction, universal serum proteinase inhibitor. Its contents
(2,5 g/l) in blood plasma is highest comparing to another proteinase
inhibitors.
The biological role of a2-macroglobulin consists in regulation of the tissue
proteolysis systems which are
very important in such physiological and pathological processes as blood
clotting, fibrinolysis, processes of immunodefence, functionality of a
complement system, inflammation, regulation of vascular tone (kinine and renin-angiothensine system).
a1-antitrypsin (a1-globulin) – glycoprotein with
a molecular weight 55 kDa. Its concentration in blood plasma is 2-3 г/л.
The main biological property of this inhibitor is its capacity to form
complexes with proteinases oppressing proteolitic activity of
such enzymes as trypsin, chemotrypsin,
plasmin, trombin. The content of a1-antitrypsin is markedly increased in inflammatory processes. The
inhibitory activity of a1-antitrypsin is very important in pancreas necrosis and acute
pancreatitis because in these conditions the proteinase level in blood and
tissues is sharply increased. The congenital deficiency of a1-antitrypsin results in the lung emphysema.
Fibronectin – glycoprotein of blood plasma that is synthesized and secreted in
intercellular space by different cells. Fibronectin present on a
surface of cells, on the basal membranes, in connective tissue and in
blood. Fibronectin has properties of a «sticking» protein and contacts with the carbohydrate groups of gangliosides on a surface
of plasma membranes executing the integrative function in intercellular interplay. Fibronectin also plays important role in the formation of the pericellular matrix.
Haptoglobin - protein of a2-globulin fraction of blood
plasma. Haptoglobin has capacity to bind a free haemoglobin forming a
complex that refer to b-globulins electrophoretic fraction. Normal
concentration in blood plasma - 0,10-0,35 g/l.
Haptoglobin-hemoglobin complexes are absorbed by the cells of reticulo-endothelial system, in
particular in a liver, and oxidized to cholic pigments. Such haptoglobin
function promotes the preservation of iron ions in an organism under conditions
of a physiological and pathological erythrocytolysis.
Transferrin - glycoprotein belonging to the b-globulin fraction. It binds in a blood plasma
iron ions (Fe3+). The protein has on the surface two centers of linkage
of iron. Transferrin is a transport form of iron delivering its to
places of accumulation and usage.
Ceruloplasmin - glycoprotein of the a2-globulin fraction. It can bind the copper ions in blood plasma. Up to 3
% of all copper contents in an organism and more than 90 % copper contents in plasma is included in ceruloplasmin. Ceruloplasmin has
properties of ferroxidase oxidizing the iron ions.
The decrease of ceruloplasmin in organism (Wilson disease) results in exit of copper ions from vessels and its accumulation in the
connective tissue that shows by pathological changes in a liver, main brain, cornea.
The place of synthesis of each fraction and subfruction of blood plasma
proteins.
Albumins,
a1-globulins,
fibrinogen are fully synthesized in hepatocytes. Immunoglobulins are produced
by plasmocytes (immune cells). In liver cryoglobulins and some other g-globulins
are produced too. a2-globulins
and b-globulins
are partly synthesized in liver and partly in reticuloendothelial cells.
Causes
and consequences of protein content changes in blood plasma.
Hypoproteinemia - decrease of
the total contents of proteins in blood plasma. This state occurs in old people
as well as in pathological states accompanying with the oppressing of protein
synthesis (liver diseases) and activation of decomposition of tissue proteins
(starvation, hard infectious diseases, state after hard trauma and operations, cancer).
Hypoproteinemia (hypoalbuminemia) also occurs in kidney diseases, when the
increased excretion of proteins via the urine takes place.
Hyperproteinemia
- increase of the
total contents of proteins in blood plasma. There are two types of hyperproteinemia - absolute and
relative.
Absolute hyperproteinemia – accumulation
of the proteins in blood. It occurs in infection and inflammatory diseases
(hyperproduction of immunoglobulins),
rheumatic diseases (hyperproduction of C-reactive protein), some
malignant tumors (myeloma) and others.
Relative hyperproteinemia – the increase of the protein concentration but not the
absolute amount of proteins. It occurs when organism loses water (diarrhea,
vomiting, fever, intensive physical activity etc.).
The
principle of the measurement of protein fractions by electrophoresis
method.
Electrophoresis
is the separation of proteins on the basis of their electric charge. It depends
ultimately on their base-acid properties, which are largely determined by the
number and types of ionizable R groups in their polipeptide chains. Since
proteins differ in amino acid composition and sequence, each protein has
distinctive acid-base properties. There are a number of different forms of
electroforesis useful for analyzing and separating mixtures of proteins
If
a precursor of an antibody-secreting cell becomes cancerous, it divides
uncontrollably to generate a clone of plasma cells
secreting a single kind of antibody molecule. The image (courtesy of Beckman
Instruments, Inc.) shows — from left to right — the electrophoretic separation
of:
1.
normal human serum with its diffuse band of gamma
globulins;
2.
serum
from a patient with multiple myeloma producing an IgG myeloma protein;
3.
serum
from a patient with Waldenström's macroglobulinemia where the cancerous
clone secretes an IgM antibody;
4.
serum
with an IgA myeloma protein.
§
Gamma
globulins can be harvested from donated blood (usually pooled from several
thousand donors) and injected into persons exposed to certain diseases such as
chicken pox and hepatitis. Because such preparations of immune globulin
contain antibodies against most common infectious diseases, the patient gains
temporary protection against the disease.
Because
of their relationship to cardiovascular disease, the analysis of serum lipids
has become an important health measure.
The
table shows the range of typical values as well as the values above (or below)
which the subject may be at increased risk of developing atherosclerosis.
·
Total
cholesterol is the serum of blood
o
HDL
cholesterol
o
LDL
cholesterol and
o
20%
of the triglyceride value
·
Note
that
o
high
LDL values are bad, but
o
high
HDL values are good.
·
Using
the various values, one can calculate a
cardiac risk ratio = total cholesterol divided by HDL
cholesterol
A
cardiac risk ratio greater than 7 is considered a warning
A.
Protein fractions which are received by the
electrophoresis
|
Fractions
|
Concentration
|
Relative
contents
|
|
Albumin
|
38,0
- 50,0 g/l
|
0,50
- 0,60
|
|
α1
globulins
|
1,4
– 3,0 g/l
|
0,01
- 0,05
|
|
α2
globulins
|
5,6
– 9,1 g/l
|
0,07
- 0,13
|
|
β-
globulins
|
5,4
– 9,1 g/l
|
0,09
– 0,15
|
|
γ
globulins
|
9,1
– 14,7 g/l
|
0,14
– 0,22
|
|
Total protein
|
65,0
– 85, 0 g/l
|
1,00
|
B.
Protein fractions which are received
with the help of imunoelectropheresis on agar gel.
|
Protein
|
Concentration
|
|
|
Acidic
α1 glycoproteid
|
|
0,20
– 0,40 g/l
|
|
α1Antitrypsyn
|
|
2,00-4,00
g/l
|
|
Ceruloplasmin
|
|
0,15-0,60
g/l
|
|
Cu2+
|
16,0-31,0
mkmmol/l
|
|
|
Haptoglobine
|
|
1,00-4,00
g/l
|
|
α2
- Macroglobulin
|
|
2,50-3,50
g/l
|
|
Transpheryn
|
|
2,50-4,10
g/l
|
|
Fe3+
|
11,0-27,0
mkmmol/l
|
|
|
Fibrinogen
|
|
2,00-4,00
g/l
|
|
Immunoglobulins
(Ig)
|
|
|
|
IgG
|
|
8,00-18,00
g/l
|
|
IgA
|
|
1,00-4,00
g/l
|
|
IgM
|
|
0,60-2,80
g/l
|
|
IgD
|
|
0,00-0,15
g/l
|
|
IgE
|
|
Till
5x10-4
|
|
|
|
|
Residual nitrogen, its components,
ways of their formation, blood content
The state of protein nutrition can be determined by measuring the
dietary intake and output of nitrogenous compounds from the body. Although
nucleic acids also contain nitrogen, protein is the major dietary source of
nitrogen and measurement of total nitrogen intake gives a good estimate of
protein intake (mg N Ч
6.25 = mg protein, as nitrogen is 16% of most proteins). The output of nitrogen
from the body is mainly in urea and smaller quantities of other compounds in
urine and undigested protein in feces, and significant amounts may also be lost
in sweat and shed skin.
The difference between intake and output of nitrogenous compounds is
known as nitrogen balance. Three states can be defined: In a healthy
adult, nitrogen balance is in equilibrium when intake equals output, and
there is no change in the total body content of protein. In a growing child, a
pregnant woman, or in recovery from protein loss, the excretion of nitrogenous
compounds is less than the dietary intake and there is net retention of
nitrogen in the body as protein, ie, positive nitrogen balance. In
response to trauma or infection or if the intake of protein is inadequate to
meet requirements there is net loss of protein nitrogen from the body, ie, negative
nitrogen balance. The continual catabolism of tissue proteins creates the
requirement for dietary protein even in an adult who is not growing, though
some of the amino acids released can be reutilized.
Nitrogen
balance studies show that the average daily requirement is 0.6 g of protein per
kilogram of body weight (the factor 0.75 should be used to allow for individual
variation), or approximately 50 g/d. Average intakes of protein in developed
countries are about 80–100 g/d, ie, 14–15% of energy intake. Because growing
children are increasing the protein in the body, they have a proportionately
greater requirement than adults and should be in positive nitrogen balance.
Even so, the need is relatively small compared with the requirement for protein
turnover. In some countries, protein intake may be inadequate to meet these
requirements, resulting in stunting of growth.
Residual nitrogen – nonprotein
nitrogen, that is nitrogen of organic and inorganic compounds that remain in
blood after protein sedimentation.
Organic
and inorganic compounds of residual nitrogen are as follows: urea (50 % of the
residual nitrogen), amino acids (25 %), creatine and creatinine (7,5 %), salts
of ammonia and indicane (0,5 %), other compounds (about 13 %).
Urea
is formed in liver during the degradation of amino acids, pyrimidine
nucleotides and other nitrogen containing compounds. Amino acids are formed as
result of protein decomposition or owing to the conversion of fatty acids or
carbohydrates to amino acids. The pool of amino acids in blood is also
supported by the process of their absorption in intestine. Creatine is produced
in kidneys and liver from amino acids glycine and arginine, creatinine is
formed in muscles as result of creatine phosphate splitting. In result of
ammonia neutralization the ammonia salts can be formed. Indicane is the product
of indol neutralization in the liver.
Creatinine Urine
The content of residual nitrogen in blood is 0,2 –
0,4 g/l.
The pathways of convertion of amino acid nonnitrogen residues.
The removal of the amino group of an
amino acid by transamination or oxidative deamination produces an α-keto
acid that contains the carbon skeleton from the amino acid (nonnitrogen residues). These α-keto
acids can be used for the biosynthesis of non-essential amino acids or
undergoes a different degradation process. For alanine and serine, the
degradation requires a single step. For most carbon arrangements, however,
multistep reaction sequences are required.
There are only seven degradation sequences for 20 amino acids. The seven
degradation products are pyruvate, acetyl CoA, acetoacetyl CoA,
α-ketoglutarate, succinyl CoA, fumarate, and oxaloacetate. The last four
products are intermediates in the citric acid cycle. Some amino acids have more
than one pathway for degradation.
The
major point of entry into the tricarboxylate cycle is via acetyl-CoA; 10 amino
acids enter by this route. Of these, six
(alanine, glycine, serine, threonine,
tryptophan and cysteine) are degraded to acetyl-CoA via pyruvate, five (phenylalanine, tyrosine, leucine, lysine,
and tryptophan) are degraded via acetoacetyl-CoA, and three (isoleucine,
leucine and tryptophan) yield
acetyl-CoA directly. Leucine and
tryptophan yield both acetoacetyl-CoA and acetyl-CoA as end products.
The
carbon skeletons of five amino acids (arginine,
histidine, glutamate, glutamine and proline) enter the tricarboxylic acid
cycle via a-ketoglutarate.
The
carbon skeletons of methionine,
isoleucine, and valine are ultimately degraded via propionyl-CoA and
methyl-malonyl-CoA to succinyl-CoA; these amino acids are thus glycogenic.
Fumarate
is formed in catabolism of phenylalanine,
aspartate and tyrosine.
Oxaloacetate
is formed in catabolism of aspartate and
asparagine. Aspartate is converted to the oxaloacetate by transamination.
Amino
acids that are degraded to citric acid cycle intermediates can serve as glucose
precursors and are called glucogenic. A glucogenic amino acid is an
amino acid whose carbon-containing degradation product(s) can be used to
produce glucose via gluconeogenesis.
Amino acids that are degraded to
acetyl CoA or acetoacetyl CoA can contribute to the formation of fatty acids or
ketone bodies and are called ketogenic. A ketogenic amino acid is an amino acid whose
carbon-containing degradation product(s) can be used to produce ketone bodies.
Amino
acids that are degraded to pyruvate can be either glucogenic or ketogenic.
Pyruvate can be metabolized to either oxaloacetate (glucogenic) or acetyl CoA
(ketogenic).
Only
two amino acids are purely ketogenic: leucine and lysine. Nine amino
acids are both glucogenic and ketogenic: those degraded to pyruvate
(alanine, glycine, cysteine, serine, threonine, tryptophan), as well as
tyrosine, phenylalanine, and isoleucine (which have two degradation products).
The remaining nine amino acids are purely glucogenic (arginine, asparagine,
aspartate, glutamine, glutamate, valine,
histidine, methionine, proline)
Clinical significance of residual nitrogen
measurement in blood. The kinds of azotemia.
Azotemia - increase of the residual nitrogen content in
blood. There are two kinds of azotemia: absolute
and relative.
Absolute azotemia – accumulation of the
components of residual nitrogen in blood. Relative
azotemia occurs in dehydration of the organism (diarrhea, vomiting).
Absolute
azotemia can be divided on the productive
azotemia and retention azotemia.
Retention azotemia is caused by the poor excretion of the nitrogen
containing compounds via the kidneys; in this case the entry of nitrogen
containing compounds into the blood is normal.
Retention azotemia can
be divided on the renal and extrarenal.
Renal retention azotemia occurs in kidney diseases (glomerulonephritis,
pyelonephritis, kidney tuberculosis et c.). Extrarenal
retention azotemia is caused by the violations of kidney hemodynamic and
decrease of glomerulus filtration processes (heart failure, local disorders of
kidney hemodynamic).
Productive azotemia is conditioned by the enhanced entry of nitrogen containing compounds into
the blood. The function of kidneys in this case doesn’t suffer. Productive
azotemia can be observed in cachexia, leukoses, malignant tumors, treatment by
glucocorticoids.
Alternate Names : Azotemia - Prerenal, Renal
Underperfusion, Uremia
Azotemia is a
medical condition characterized by abnormal levels of urea, creatinine, various body waste
compounds, and other nitrogen-rich compounds in the blood as a result of insufficient
filtering of the blood by the kidneys.
Uremia can be used as a synonym, or can
be used to indicate severe azotemia, in which symptoms are produced.
Azotemia
can be classified according to its cause. In prerenal azotemia the blood
supply to the kidneys is inadequate. In postrenal azotemia the urinary
outflow tract is obstructed. Other forms of azotemia are caused by diseases of
the kidneys themselves.
Other
causes of azotemia include congestive heart failure, shock, severe
burns, prolonged vomiting or diarrhea, some antiviral medications, liver
failure, or trauma to the kidney(s).
·
Thirst,
swelling (edema,
anasarca)
·
Orthostatic
blood pressure (rises or falls, significantly depending on position)
A urinalysis will typically show a decreased
urine sodium level, a high urine creatinine-to- serum creatinine ratio, a high
urine urea-to-serum urea ratio, and concentrated urine (determined by
osmolality and specific gravity). None of these is particularly useful in
diagnosis.
Prompt
treatment of some causes of azotemia can result in restoration of kidney
function; delayed treatment may result in permanent loss of renal function.
Treatment may include hemodialysis or peritoneal dialysis, medications to increase
cardiac output and increase blood pressure, and the treatment of the condition
that caused the azotemia to begin with. NOTE: Azotemia is not diagnosed with
abnormally high levels of Creatinine. Azotemia simply refers to an elevated
level of urea in the blood.
Added
Note: Uremia is not azotemia. Azotemia is one of many
clinical characteristics of uremia, which is a syndome characteristic of renal
disease. Uremia includes Azotemia, as well as acidosis, hyperkalemia,
hypertension, anemia and hypocalcemia along with other findings.
Lipoproteins and Apoproteins
http://www.youtube.com/watch?v=97uiV4RiSAY
Lipids are a group of fatty substances that
includes triglycerides (fat), phospholipids and sterols (e.g.
cholesterol). They constitute an important source of energy, serve as
precursors for a number of essential compounds, and are key components of cells
and tissues. Cholesterol, for example, is an indispensable constituent of
cellular membranes (1), as well as the precursor for both steroid
hormones and bile acids. On average, the body utilizes approximately 1000
milligrams of cholesterol per day, 30% of which comes directly from foods of
animal origin, and the rest is synthesized in the liver. Due to the
insolubility of cholesterol and other fatty compounds in the blood, their
redistribution in the body requires specialized carriers capable of
solubilzing, ferrying, and unloading them at specific target sites. Miscarriage
of lipids while in circulation may lead to atherosclerosis; a clinical
condition marked by fatty deposits in the inner walls of arteries, and the
leading cause of death and disability in Western countries.
Most lipids are transported in the blood as part
of soluble complexes called lipoproteins (LPs). Plasma LPs are spherical
particles composed of a hydrophobic lipid core surrounded by a hydrophilic
layer, which renders the particles soluble. The lipid core contains primarily
triglycerides (TG) and cholesteryl esters (CE), as well as small amounts of
other fatty compounds, such as sphingolipids and fat-soluble vitamins (e.g.
vitamins A, D, E, and K). The external layer is made of phospholipids,
unesterified cholesterol, and specialized proteins, called apolipoproteins or
apoproteins. These proteins facilitate lipid solubilization and help to
maintain the structural integrity of LPs. They also serve as ligands for LP
receptors and regulate the activity of LP metabolic enzymes. As depicted
in (Figure 1), the amphipathic molecules that compose the
outer layer of LPs are arranged so that their hydrophobic parts face the
central core, and their hydrophilic regions face the surrounding aqueous
environment.
Figure 1: Schematic Illustration of a
Lipoprotein Particle
Cholesteryl esters, which do not contain a free
hydroxyl group (-OH) are more hydrophobic than cholesterol, and better
accommodated in the core of LPs. The conversion of cholesterol to CE is
catalyzed by a LP-associated enzyme called lecithin-cholesterol acyltransferase
(LCAT). This enzyme, which promotes packaging of cholesteryl molecules in LPs,
is critical for normal cholesterol metabolism. Deficiency of LCAT activity
leads to accumulation of unesterified cholesterol in tissues, and is associated
with a number of clinical conditions including corneal opacity, hemolytic
anemia, and premature atherosclerosis.
During ordinary metabolism, plasma LPs lose,
acquire, and exchange their lipid and protein constituents. Normally, fat-rich
LPs lose most of their fat within a few hours of food ingestion, and become
smaller and denser particles with higher relative cholesterol content. The
depletion of fat from LPs is catalyzed by lipoprotein lipase (LPL). This
lipolytic enzyme is located on the surface of endothelial capillaries, and
degrades triglycerides to free fatty acids (FFAs) and glycerol. The released
FFAs may stay in circulation bound to albumin, or be taken-up by muscle and fat
cells for usage and storage, respectively.
Lipids of dietary origin are processed by
intestinal epithelial cells, and then secreted into the bloodstream as part of
large, fat-rich LPs called chylomicrons (chylo = milky, micron= indicates
particle size). En route to the liver, chylomicrons (CM) pass through
endothelial capillaries, lose some fat, and their remnants are taken-up by
liver cells. In the liver, the lipids obtained from CM remnants are
re-processed and then secreted back into the bloodstream as part of very
low-density LPs (VLDL). Depletion of fat from VLDL transforms the particle into
an intermediate density lipoprotein (IDL), which upon further degradation of
its fat is converted into a relatively stable particle, called low density
lipoprotein (LDL). Because of its high cholesterol content, LDL is also called
LDL-cholesterol. Of the total blood cholesterol, 60-75% is found in LDL and the
rest primarily in high-density lipoprotein (HDL) particles. The main
characteristics of plasma LPs and their associated apoproteins are summarized
in (Tables I and II),
respectively.
All peripheral cells express the LDL-receptor
(LDLR), and recycle it to the cell surface upon need for cholesterol.
Cholesterol is delivered to these cells through binding of LDL to LDLR, which
triggers endocytosis (internalization) of both species. When the need for
cholesterol is satisfied, the recycling of LDLR is discontinued.
Normally, an LDL particle stays in circulation for no more than a few days
before being consumed by a cholesterol needing cell. However, under conditions
of sustained cholesterol excess, the particle stays in circulation for longer
periods of time, and becomes more vulnerable to undesired modifications (e.g.
oxidation). As high levels of oxidized LDL are commonly found in
atherosclerotic plaques, they are thought to be the major inducer of atherosclerotic
lesions. Hence, LDL became known as bad cholesterol. However, today we know
that not all LDL particles are bad, and that some LDL particles, especially
very large ones (with diameter >21.3nm), may even provide protection against
atherosclerosis (2). LDL and HDL particle sizes are largely
determined by a LP-associated protein, called CETP (cholesteryl ester transfer
protein). This protein enhances exchange of non-polar lipids, primarily CE and
TG, and facilitates tight packaging of CE within the core of the particles. The
end result of prolonged and/or efficient CETP action is smaller LDL and HDL
particles. [The LP-anchored CETP can be envisioned as having a hand that
rotates between the interior and exterior of the particle and capable of
holding only one lipid molecule at a time. Grasping of one molecule releases
another and vise versa.]
Genetic variation at the human CETP gene
generates proteins with varying degrees of activity. For example, a
single codon variation, from isoleucine to valine at position 405, generates a
mutant protein, designated I405V, which manifests significantly reduced CETP
activity (3, 4). In a new observational study, Barzilai, N. et
al. (2) found that people with homozygosity for
the I405V allele have larger HDL and LDL particles, and that this genotype is associated
with exceptional longevity and a markedly reduced risk of coronary artery
disease (CAD). Of the 213 centenarians enrolled in the study, 80% had a high
proportion of large LDL particles, compared to just 8% of the subjects in the
control group (256 people in their 60’s and 70’s) (2).
Interestingly, HDL and LDL particle sizes are significantly larger in women
than in men, which may account, at least in part, for the longer life
expectancies of women.
Unlike LDL, HDL is not recognized by LDLR, and
cannot deliver cholesterol to tissue cells. Instead, it has the ability to
remove excess peripheral cholesterol and return it to the liver for recycling
and excretion. This process, called reverse cholesterol transport, is thought
to protect against atherosclerosis. Observational studies over the last 2
decades have consistently shown strong correlation between elevated HDL levels
and low incidents of coronary heart disease (CHD). Hence HDL has been dubbed
“good” cholesterol.
HDL is synthesized in the liver and intestine as
a nascent, discoid-shaped particle that contains predominantly apoA-I, and some
phospholipids. Upon maturation, HDL assumes a spherical shape, and the
composition of its core lipids becomes very similar to that of LDL. However,
the relative higher protein content in HDL renders the particle denser and more
resistant to undesired modifications. Unlike the case of LDL, the clearance of
HDL from circulation is not negatively affected by excess cholesterol, which
may be another reason why HDL, despite being much smaller particle than LDL
(10nm versus 20nm), is not found in atherosclerotic plaques. It’s worth noting,
that the potential of LPs to become harmful is also influenced by the character
of their lipid constituents. For example, vitamin E and lipids containing
omega-3 fatty acid moieties appear to protect the particles from harmful
oxidation and from getting stuck on the walls of blood vessels.
The functional difference between LDL and HDL
results primarily from the different character of their major apoproteins,
apoB-100 and apoA-I, respectively. ApoB-100, which is found in VLDL, IDL,
and LDL, but not in HDL, serves as a ligand for LDLR, and provides LDL with the
means to deliver cholesterol to tissue cells. On the other hand, apoA-I, which
is found exclusively in HDL, has a unique ability to capture and solubilze free
cholesterol. This apoA-I ability enables HDL to act as a cholesterol scavenger.
A mutant apoA-I protein, called apoA-I Milano
(apoA-Im), has been identified in a group of people that live in a small
village in northern Italy (5). Carriers of this protein, all heterozygous for
the mutation, had very low levels of HDL (7-14 mg/dl) but showed no clinical
signs of atherosclerosis (5-7). HDL particles in these subjects were markedly
larger than control (12nm versus 9.4nm), which may account for their immunity
against premature atherosclerosis. ApoA-Im differs from natural apoA-I by
having a cysteine residue at position 173 instead of arginine. This cysteine
residue forms disulfide bridges with other apoA-I molecules or with apoA-II (6, 7), which apparently lead to larger HDL particles. It also renders apoA-I
more susceptible to catabolism (8), accounting for the
low HDL levels in apoA-Im carriers.
The therapeutic potential of apoA-I has been
recently assessed in patients with acute coronary syndromes (9). Of the 47 patients that participated in a randomized controlled trial,
36 received 5 weekly infusions of recombinant apoA-Im/phospholipid complexes,
and 11 received only saline infusions. The results showed significant
regression in coronary atherosclerotic volume in the apoA-Im treated group, and
virtually no change in the control group (9).
These results, if reproduced in larger clinical trials, may constitute a
revolutionary breakthrough in the non-invasive treatment of cardiovascular
disease. They should also encourage further exploration into the therapeutic
usefulness of apoA-Im and normal apoA-I in managing atherosclerotic vascular
diseases.
lipoproteins).
The lilipoproteins - Any of the series of soluble lipid-protein
complexes which are transported in the blood; each aggregate particle consists
of a spherical hydrophobic core containing triglycerides and cholesterol esters
surrounded by an amphipathic monolayer of phopholipids, cholesterol and
apolipoproteins; classes of lipoproteins include chylomicrons, very low-density
lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density
lipoproteins (LDL), and high-density lipoproteins (HDL).
chylomicrons -
The class of largest diameter soluble lipid-protein complexes which the lowest
in density (mass to volume ratio); their composition is ~2% apolipoproteins,
~5% cholesterol, and ~93% triglycerides and phospholipids; their normal role is
to be synthesized by the intestinal mucosal cells to transport dietary
(exogenous) triglycerides and other lipids from the intestines via the lacteals
and lymphatic system to the systemic circulation to the adipose tissue and liver
for storage and use; they are only present in the blood in significant
quantities after the digestion of a meal.
low-density
lipoproteins (LDL) - The class of large diameter
soluble lipid-protein complexes which the fourth lowest in density (mass to
volume ratio); their composition is ~25% apolipoproteins, ~45% cholesterol, and
~30% triglycerides and phospholipids; their normal role is to transport
cholesterol and other lipids from the liver and intestines to the tissues for
use; elevated levels of LDL are associated with increased risk of
cardiovascular disease. nickname - bad cholesterol
high-density
lipoproteins (HDL) - The class of small diameter
soluble lipid-protein complexes which the highest in density (mass to volume
ratio); their composition is ~45% apolipoproteins, ~25% cholesterol, and ~30%
triglycerides and phospholipids; their normal role is to transport cholesterol
and other lipids from the tissues to the liver for disposal; elevated levels of
HDL are associated with decreased risk of cardiovascular disease.
very
low-density lipoproteins (VLDL) - The class of very large
diameter soluble lipid-protein complexes which the second lowest in density
(mass to volume ratio); their composition is ~10% apolipoproteins, ~40%
cholesterol, and ~50% triglycerides and phospholipids; their normal role is to
transport triglycerides and other lipids from the liver and intestines to the
tissues for use; elevated levels of VLDL are associated with some increased
risk of cardiovascular disease.
formation of lipoproteins
http://www.youtube.com/watch?v=97uiV4RiSAY


{kind=link}
{kind=link}
{kind=link}



![Hemoglobin also carries nitric oxide in the globin part of the molecule. This improves oxygen delivery in the periphery and contributes to the control of respiration. NO binds reversibly to a specific cystein residue in globin; the binding depends on the state (R or T) of the hemoglobin. The resulting S-nitrosylated hemoglobin influences various NO-related activities such as the control of vascular resistance, blood pressure and respiration. NO is released not in the cytoplasm of erythrocytes but is transported by an anion exchanger called AE1 out of them.[]](http://themedicalbiochemistrypage.org/images/bohr.jpg){kind=link}
{kind=link}
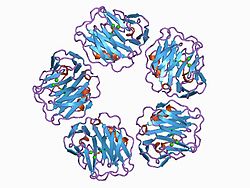{kind=link}
{kind=link}