Basic principles of
metabolism: catabolism, anabolism. Common pathways of proteins, carbohydrates
and lipids transformation. Studying of Krebs cycle functioning. Bioenergetics
processes: biological oxidation, oxidative phosphorylation, ATP synthesis.
BIOMEDICAL IMPORTANCE
The fate of dietary components after
digestion and absorption constitutes
metabolism—the metabolic pathways taken by
individual molecules, their interrelationships, and the mechanisms that regulate the flow of metabolites through the pathways. Metabolic pathways fall into three categories: (1) Anabolic pathways are those involved in the synthesis of compounds. Protein synthesis is such a pathway, as is the synthesis of
fuel reserves of triacylglycerol and
glycogen. Anabolic pathways are
endergonic. (2) Catabolic pathways are involved in the breakdown of larger molecules, commonly involving oxidative reactions; they are exergonic,
producing reducing equivalents and,
mainly via the respiratory chain, ATP.
(3) Amphibolic pathways occur at the
“crossroads” of metabolism, acting as links between the anabolic and catabolic pathways, eg, the citric acid cycle.
A knowledge of normal metabolism is
essential for an understanding of abnormalities
underlying disease. Normal metabolism includes adaptation
to periods of starvation, exercise, pregnancy, and
lactation. Abnormal metabolism may result from
nutritional deficiency, enzyme
deficiency, abnormal secretion of hormones, or
the actions of drugs and toxins. An
important example of a metabolic disease is diabetes
mellitus.
PATHWAYS THAT PROCESS THE MAJOR PRODUCTS OF DIGESTION
The nature of the diet sets the
basic pattern of metabolism. There is a
need to process the products of digestion of dietary carbohydrate, lipid, and protein. These are mainly glucose, fatty acids and glycerol, and
amino acids, respectively. In ruminants
(and to a lesser extent in other
herbivores), dietary cellulose is fermented by symbiotic microorganisms to short-chain fatty acids (acetic, propionic, butyric), and metabolism in these animals is adapted to use these fatty acids as major
substrates.
All the products of digestion are
metabolized to a common product, acetyl-CoA, which
is then oxidized by the citric acid cycle .
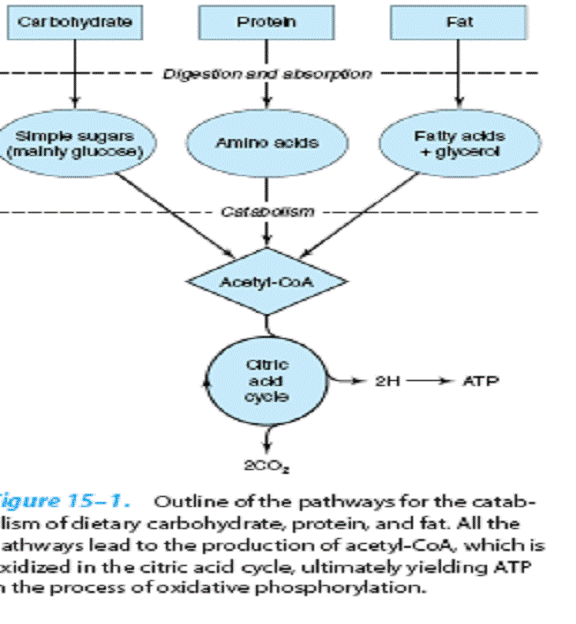
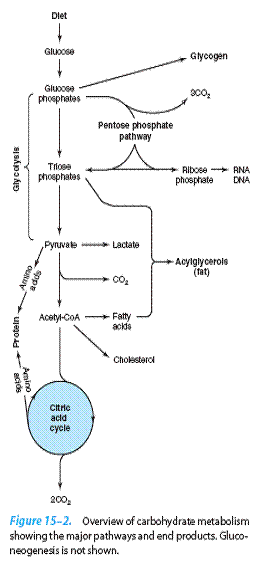
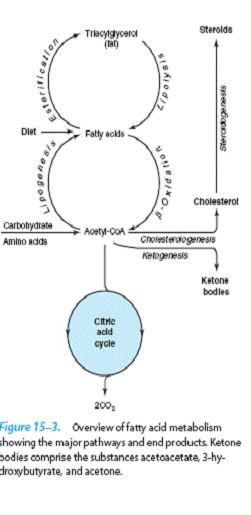
Carbohydrate Metabolism Is Centered on the Provision & Fate of Glucose
Glucose is metabolized to pyruvate by the pathway of glycolysis, which can occur anaerobically (in the absence of oxygen), when the end product is lactate. Aerobic
tissues metabolize pyruvate to acetyl-CoA,
which can enter the citric acid cycle for
complete oxidation to CO2 and H2O, linked to the
formation of ATP.
Glucose and its metabolites also
take part in other processes. Examples: (1) Conversion
to the storage polymer glycogen in skeletal
muscle and liver. (2) The
pentose phosphate pathway, an alternative to part of the pathway of glycolysis, is a source of reducing
equivalents (NADPH) for biosynthesis and the
source of ribose
for nucleotide and nucleic acid
synthesis. (3) Triose phosphate gives rise to
the glycerol moiety of
triacylglycerols. (4) Pyruvate and intermediates of the citric acid cycle provide the carbon skeletons for the synthesis of amino acids; and acetyl-CoA,
the precursor of fatty acids and cholesterol
(and hence of all steroids
synthesized in the body). Gluconeogenesis is the process of forming glucose from noncarbohydrate
precursors, eg, lactate, amino
acids, and glycerol.
Lipid Metabolism Is Concerned Mainly With Fatty Acids & Cholesterol
The source of long-chain fatty acids
is either dietary lipid or de novo synthesis from
acetyl-CoA derived from carbohydrate.
Fatty acids may be oxidized to acetyl- CoA (β-oxidation) or esterified with
glycerol, forming triacylglycerol (fat) as the body’s main fuel
reserve. Acetyl-CoA formed by β-oxidation may undergo several fates:
(1) As with acetyl-CoA arising from glycolysis, it is oxidized to CO2 + H2O via the citric acid cycle.
(2) It is the precursor for synthesis of cholesterol and other steroids.
(3) In the liver, it forms ketone bodies (acetone,
acetoacetate, and 3 hydroxybutyrate) that are important fuels in prolonged starvation.
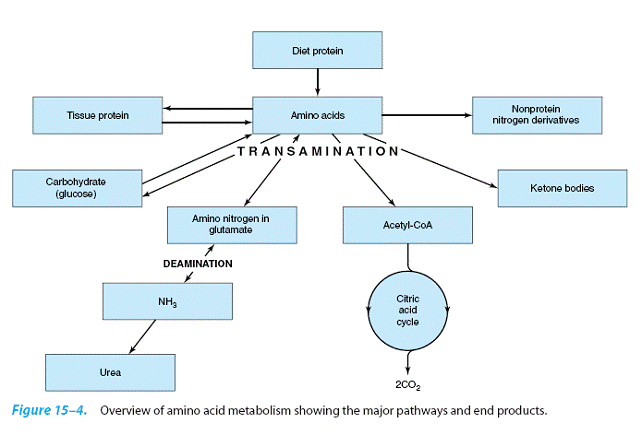
Much of Amino Acid Metabolism Involves Transamination
The amino acids are required for
protein synthesis. Some must be supplied in the diet
(the essential amino acids) since they cannot be synthesized
in the body. The remainder are nonessential
amino acids that are supplied in
the diet but can be formed from metabolic intermediates by transamination, using the amino nitrogen
from other amino acids. After deamination, amino nitrogen is excreted as urea, and the
carbon skeletons that remain after
transamination (1) are oxidized to CO2 via
the citric acid cycle, (2) form glucose (gluconeogenesis), or (3) form ketone bodies.
Several amino acids are also the
precursors of other compounds, eg, purines, pyrimidines,
hormones such as epinephrine and thyroxine, and
neurotransmitters.
METABOLIC PATHWAYS MAY BE STUDIED AT DIFFERENT LEVELS
OF ORGANIZATION
In addition to studies in the whole
organism, the location and
integration of metabolic pathways is revealed by studies at several levels of organization. At the tissue
and organ level, the nature of the substrates
entering and metabolites leaving tissues and
organs is defined. At the subcellular
level, each cell organelle (eg, the mitochondrion) or compartment (eg, the cytosol) has specific roles that form part of a subcellular pattern of metabolic pathways.
At the Tissue and Organ Level, the Blood Circulation Integrates Metabolism
Amino acids resulting from the digestion of
dietary protein and glucose resulting from
the digestion of carbohydrate are absorbed
and directed to the liver via the
hepatic portal vein. The liver has the role of regulating the blood concentration of most water-soluble
metabolites In the case of glucose, this is achieved by taking up glucose in excess of immediate requirements and converting it to glycogen.
Between meals, the liver acts to maintain the blood glucose concentration from
glycogen (glycogenolysis) and,
together with the kidney, by
converting noncarbohydrate metabolites such as lactate, glycerol, and amino acids to glucose (gluconeogenesis). Maintenance of an adequate concentration of blood glucose is vital for those tissues in which
it is the major fuel (the brain) or the only fuel (the
erythrocytes).
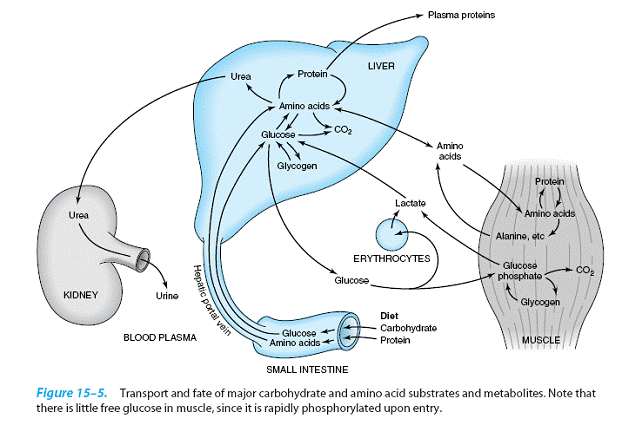
The liver also synthesizes the
major plasma proteins (eg, albumin) and deaminates
amino acids that are in excess of requirements,
forming urea, which
is transported to the kidney and
excreted. Skeletal muscle utilizes glucose as a fuel, forming both lactate and CO2. It stores glycogen as a fuel for
its use in muscular contraction and synthesizes muscle protein from plasma amino acids. Muscle accounts for approximately 50% of body mass and consequently represents a considerable store of protein that can be drawn upon to supply amino acids for gluconeogenesis in starvation.
Lipids in the diet are mainly triacylglycerol and are hydrolyzed to monoacylglycerols and fatty acids in the gut, then reesterified in the
intestinal mucosa. Here they are packaged with
protein and secreted into the lymphatic system and thence
into the
blood stream as chylomicrons, the
largest of the plasma lipoproteins.
Chylomicrons
also contain other lipidsoluble nutrients,
eg, vitamins. Unlike glucose and amino acids,
chylomicron triacylglycerol is not taken up directly by the liver. It is first metabolized by tissues that have lipoprotein lipase, which hydrolyzes the
triacylglycerol, releasing fatty acids that are
incorporated into tissue lipids or oxidized as fuel.
The other major source of
long-chain fatty acid is synthesis (lipogenesis) from carbohydrate, mainly in adipose tissue and the liver. Adipose tissue triacylglycerol is the main fuel
reserve of the body. On hydrolysis (lipolysis)
free fatty acids are released
into the circulation. These are taken up by most
tissues (but not brain or
erythrocytes) and esterified to acylglycerols
or oxidized as a fuel. In the liver, triacylglycerol arising from lipogenesis, free fatty acids, and
chylomicron remnants is secreted into the circulation as very low density lipoprotein (VLDL). This triacylglycerol undergoes a fate similar to that of chylomicrons. Partial oxidation of fatty acids in the liver leads to ketone body production
Ketone bodies are transported to extrahepatictissues, where they act as a fuel
source in starvation.
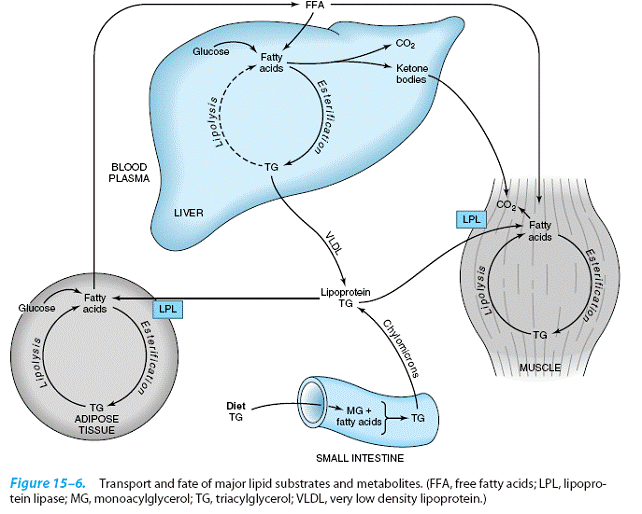
At the Subcellular Level, Glycolysis Occurs in the Cytosol & the Citric Acid Cycle in the Mitochondria
Compartmentation of pathways in separate subcellular compartments or organelles permits integration and regulation of metabolism. Not all pathways are of
equal
importance in all cells. Depicts the subcellular compartmentation of metabolic pathways in a hepatic parenchymal cell.
The central role of the mitochondrion
is immediately apparent, since it acts as the focus
of carbohydrate, lipid, and amino acid metabolism. It
contains the enzymes
of the citric acid cycle, â-oxidation of fatty acids, and ketogenesis, as well as the respiratory chain and ATP synthase. Glycolysis,
the pentose phosphate pathway, and fatty acid synthesis are all found in the cytosol. In gluconeogenesis, substrates such as lactate and pyruvate, which are formed in the cytosol, enter the mitochondrion to
yield oxaloacetate before
formation of glucose. The
membranes of the endoplasmic reticulum contain the enzyme system for acylglycerol synthesis, and the ribosomes are responsible for protein
synthesis.
SUMMARY
• The products of digestion provide
the tissues with the building blocks for the
biosynthesis of complex molecules
and also with the fuel to power the living
processes.
• Nearly all products of digestion
of carbohydrate, fat, and protein
are metabolized to a common metabolite, acetyl-CoA, before final oxidation to CO2 in the
citric acid cycle.
• Acetyl-CoA is also used as the
precursor for biosynthesis of
long-chain fatty acids; steroids, including cholesterol; and ketone bodies.
• Glucose provides carbon skeletons
for the glycerol moiety of fat and of several
nonessential amino acids.
• Water-soluble products of
digestion are transported directly to
the liver via the hepatic portal vein. The liver regulates the blood concentrations of glucose
and amino acids.
• Pathways are compartmentalized
within the cell. Glycolysis, glycogenesis,
glycogenolysis, the pentose phosphate
pathway, and lipogenesis occur in the cytosol.
The mitochondrion contains the
enzymes of the citric acid cycle, β-oxidation of fatty acids, and of oxidative phosphorylation. The endoplasmic reticulum also contains the enzymes for many other processes, including protein synthesis, glycerolipid
formation, and drug metabolism.
• Metabolic pathways are regulated
by rapid mechanisms affecting the activity of existing
enzymes, eg, allosteric and covalent modification
(often in response
to hormone action); and slow
mechanisms affecting the synthesis of enzymes.
BIOMEDICAL IMPORTANCE OF KREBS CYCLE
The citric acid cycle (
tricarboxylic acid cycle) is a series of reactions in
mitochondria that oxidize acetyl
residues (as acetyl-CoA) and reduce coenzymes that upon reoxidation are linked to the formation of ATP.
The citric acid cycle is the final
common pathway for the aerobic oxidation of
carbohydrate, lipid, and protein
because glucose, fatty acids, and most amino
acids are metabolized to acetyl-CoA
or intermediates of the cycle. It also has a central
role in gluconeogenesis, lipogenesis,
and interconversion of amino acids. Many
of these processes occur in most
tissues, but the liver is the only
tissue in which all occur to a significant extent.
The repercussions are therefore
profound when, for example, large
numbers of hepatic cells are damaged as in acute hepatitis or replaced by connective tissue (as in
cirrhosis). Very few, if any, genetic
abnormalities of citric acid cycle enzymes have been
reported; such abnormalities would be
incompatible with life or normal
development.
THE CITRIC ACID CYCLE PROVIDES SUBSTRATE FOR THE
RESPIRATORY CHAIN
The cycle starts with reaction
between the acetyl moiety of
acetyl-CoA and the four-carbon dicarboxylic acid oxaloacetate, forming a six-carbon tricarboxylic acid, citrate. In the subsequent reactions, two molecules of CO2 are released and oxaloacetate is regenerated.
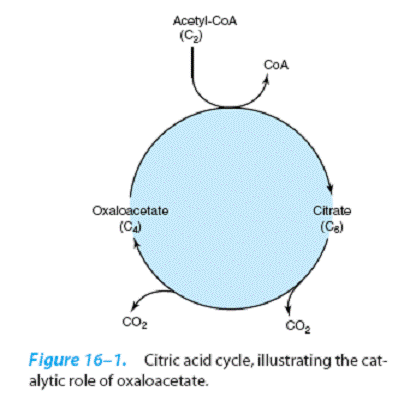
Only a small quantity of
oxaloacetate is needed for the
oxidation of a large quantity of acetyl-CoA; oxaloacetate
may be considered to play a catalytic
role. The citric acid cycle is an integral
part of the process by which much
of the free energy liberated during the oxidation of fuels is made available. During oxidation of acetyl-CoA, coenzymes are reduced and subsequently reoxidized in the respiratory chain, linked to the
formation
of ATP. This process is aerobic, requiring oxygen as the final oxidant of the reduced coenzymes. The enzymes of the citric acid cycle are
located in the mitochondrial matrix, either free or attached to the inner mitochondrial membrane, where the enzymes of the respiratory chain are also found.
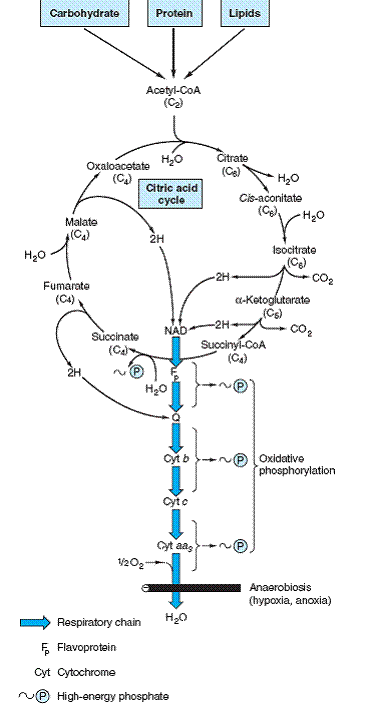
REACTIONS OF THE CITRIC ACID CYCLE LIBERATE REDUCING
EQUIVALENTS & CO2
The initial reaction between
acetyl-CoA and oxaloacetate to form
citrate is catalyzed by citrate synthase which forms a carbon-carbon bond between the methyl carbon of acetyl-CoA and the carbonyl carbon of
oxaloacetate.
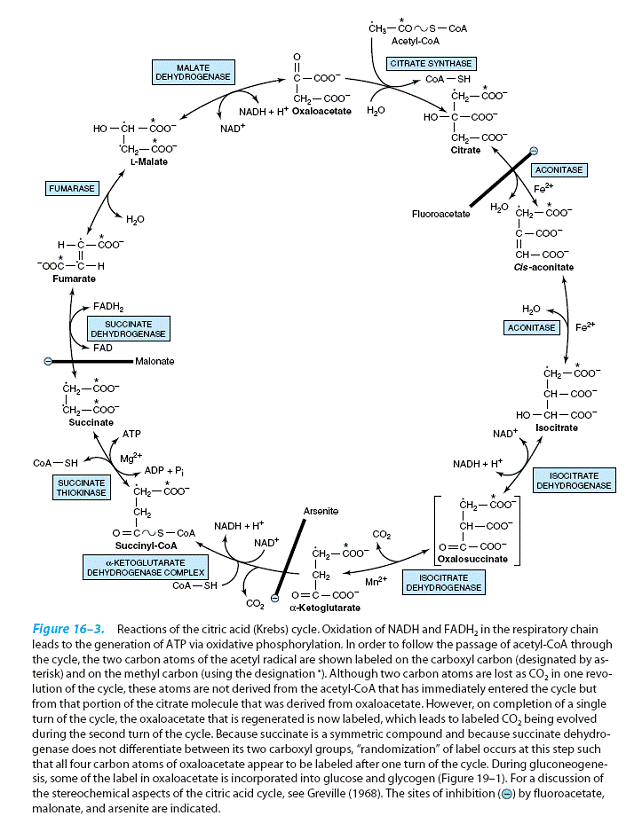
The thioester bond of the resultant citryl-CoA is hydrolyzed, releasing
citrate and CoASH—an exergonic reaction. Citrate is isomerized to isocitrate by the enzyme
aconitase (aconitate hydratase); the reaction
occurs in two steps: dehydration to cis-aconitate,
some of which remains bound to the enzyme; and
rehydration to isocitrate.
Although citrate is a symmetric
molecule, aconitase reacts with citrate asymmetrically,
so that the two carbon atoms that are lost in
subsequent reactions of
the cycle are not those that were
added from acetyl- CoA. This asymmetric behavior is due
to channeling— transfer of
the product of citrate synthase directly onto
the active site of aconitase without
entering free solution. This
provides integration of citric acid cycle activity and the provision of citrate in the cytosol as a source
of acetyl-CoA for fatty acid
synthesis. The poison fluoroacetate is toxic because fluoroacetyl-CoA condenses with oxaloacetate to form fluorocitrate, which inhibits
aconitase, causing citrate to
accumulate. Isocitrate undergoes dehydrogenation
catalyzed by isocitrate dehydrogenase to form, initially, oxalosuccinate,
which remains enzyme-bound and
undergoes decarboxylation to α-ketoglutarate. The decarboxylation requires Mg2+ or Mn2+ ions.
There are three isoenzymes of
isocitrate dehydrogenase. One, which uses NAD+, is found only in mitochondria. The other two use NADP+ and are found in mitochondria and the cytosol.
Respiratory
chain-linked oxidation of isocitrate
proceeds almost completely through the
NAD+-dependent enzyme. α-Ketoglutarate undergoes oxidative decarboxylation
in a reaction catalyzed by a
multi-enzyme complex similar to that involved in the
oxidative decarboxylation of pyruvate.
The ketoglutarate dehydrogenase complex requires the same cofactors as the pyruvate dehydrogenase complex—thiamin diphosphate, lipoate, NAD+, FAD, and CoA—and results in the formation of succinyl-CoA. The equilibrium of this reaction is so much in favor of succinyl-CoA formation that it must be considered physiologically
unidirectional.
As in the case of pyruvate
oxidation, arsenite inhibits the reaction, causing the substrate ketoglutarate, to accumulate. Succinyl-CoA is converted to succinate by the enzyme succinate thiokinase (succinyl-CoA synthetase).
This is the only example in the
citric acid cycle of substrate-level phosphorylation.
Tissues in which gluconeogenesis occurs (the
liver and kidney) contain two
isoenzymes of succinate thiokinase,
one specific for GDP and the other for ADP. The GTP
formed is used for the decarboxylation of
oxaloacetate to phosphoenolpyruvate
in gluconeogenesis and provides a regulatory link between citric acid cycle activity and the withdrawal of oxaloacetate for gluconeogenesis. Nongluconeogenic tissues have only the isoenzyme that uses ADP.
When ketone bodies are being
metabolized in extrahepatic tissues
there is an alternative reaction catalyzed by succinyl-CoA–acetoacetate-CoA transferase (thiophorase)— involving transfer of CoA from succinyl- CoA to acetoacetate, forming acetoacetyl-CoA. The onward metabolism of succinate, leading to the regeneration of oxaloacetate, is the same sequence of chemical reactions as occurs in the β-oxidation of fatty acids: dehydrogenation to form a carbon-carbon double
bond, addition of water to form a
hydroxyl group, and a further dehydrogenation to yield
the oxo- group of oxaloacetate.
The first dehydrogenation reaction,
forming fumarate, is catalyzed by succinate dehydrogenase,
which is bound to the inner surface of the
inner mitochondrial
membrane. The enzyme contains FAD
and iron-sulfur (Fe:S) protein and directly reduces
ubiquinone in the respiratory chain. Fumarase
(fumarate hydratase) catalyzes the addition
of water across the double bond of fumarate,
yielding malate. Malate is converted to oxaloacetate by malate dehydrogenase, a reaction requiring
NAD+. Although the equilibrium of
this reaction strongly favors malate, the net flux
is toward the direction of oxaloacetate
because of the continual removal of
oxaloacetate (either to form
citrate, as a substrate for gluconeogenesis,
or to undergo transamination to aspartate) and also because of the continual reoxidation of NADH.
TWELVE ATP ARE FORMED PER TURN OF THE CITRIC ACID CYCLE
As a result of oxidations catalyzed
by the dehydrogenases of the
citric acid cycle, three molecules of NADH and one of FADH2 are produced for each molecule of
acetyl-CoA catabolized in one turn
of the cycle. These reducing equivalents are transferred
to the respiratory chain, where reoxidation of each
NADH results in formation of 3 ATP and
reoxidation of FADH2 in formation of 2 ATP. In
addition, 1 ATP (or GTP) is formed by
substrate-level phosphorylation catalyzed by
succinate thiokinase.
VITAMINS PLAY KEY ROLES IN THE
CITRIC ACID CYCLE
Four of the B vitamins are essential
in the citric acid cycle and therefore in
energy-yielding metabolism: (1) riboflavin, in the form of flavin adenine
dinucleotide
(FAD), a cofactor in the α-ketoglutarate dehydrogenase complex and in succinate dehydrogenase; (2) niacin,
in the form of nicotinamide adenine
dinucleotide (NAD),
the coenzyme for three
dehydrogenases in the cycle— isocitrate
dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase; (3) thiamin (vitamin
B1), as thiamin diphosphate, the coenzyme for
decarboxylation in the α-ketoglutarate dehydrogenase
reaction; and (4) pantothenic acid, as
part of coenzyme A,
the cofactor attached to “active”
carboxylic acid residues such as
acetyl-CoA and succinyl-CoA.
THE CITRIC ACID CYCLE PLAYS A PIVOTAL ROLE IN METABOLISM
The citric acid cycle is not only a
pathway for oxidation of
two-carbon units—it is also a major pathway for interconversion of metabolites arising from transamination
and deamination of amino
acids. It also provides the
substrates for amino acid synthesis by transamination, as well as for gluconeogenesis and fatty
acid synthesis.
Because it functions in both
oxidative and synthetic processes,
it is amphibolic.
The Citric Acid Cycle Takes Part in Gluconeogenesis, Transamination, & Deamination
All the intermediates of the cycle
are potentially glucogenic, since they
can give rise to oxaloacetate and thus net
production of glucose (in the liver and kidney, the
organs that carry out
gluconeogenesis). The key enzyme that catalyzes net
transfer out of the cycle into gluconeogenesis is phosphoenolpyruvate carboxykinase, which decarboxylates oxaloacetate to phosphoenolpyruvate, with GTP acting as the donor phosphate. Net transfer
into the cycle occurs as a result of several different reactions. Among the most important of such anaplerotic reactions is the formation of oxaloacetate by the carboxylation of pyruvate, catalyzed by pyruvate carboxylase. This reaction is important in maintaining an adequate concentration of oxaloacetate for the condensation reaction with acetyl-CoA. If
acetyl- CoA accumulates, it acts both as an
allosteric activator of pyruvate carboxylase and as an
inhibitor of pyruvate dehydrogenase,
thereby ensuring a supply of oxaloacetate.
Lactate, an important substrate for
gluconeogenesis, enters the cycle via oxidation to
pyruvate and then carboxylation to oxaloacetate.
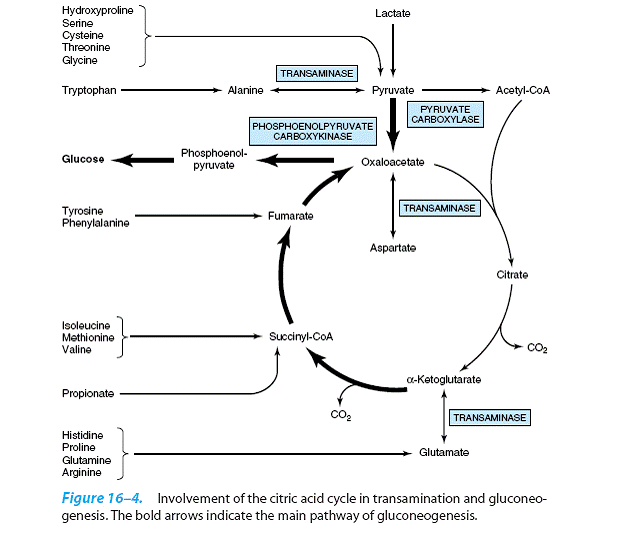
Aminotransferase (transaminase) reactions form pyruvate from alanine, oxaloacetate from aspartate,
and α-ketoglutarate from glutamate.
Because these reactions are
reversible, the cycle also serves as a source of carbon skeletons for the synthesis of these amino acids. Other amino acids contribute to gluconeogenesis
because their carbon skeletons give rise to
citric acid cycle intermediates. Alanine, cysteine,
glycine, hydroxyproline, serine,
threonine, and tryptophan yield pyruvate;
arginine, histidine, glutamine, and
proline yield α-ketoglutarate; isoleucine,
methionine, and valine yield succinyl-CoA;
and tyrosine and phenylalanine yield fumarate.
In ruminants, whose main metabolic
fuel is shortchain fatty acids formed by bacterial
fermentation, the conversion of propionate, the major
glucogenic product of rumen fermentation, to succinyl-CoA
via the methylmalonyl-CoA pathway is
especially
important.
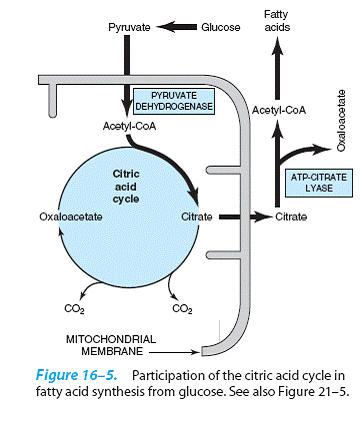
The Citric Acid Cycle Takes Part in Fatty Acid Synthesis
Acetyl-CoA, formed from pyruvate by
the action of pyruvate dehydrogenase, is the major
building block for long-chain fatty acid synthesis in
nonruminants. (In ruminants, acetyl-CoA
is derived directly from acetate.)
Pyruvate dehydrogenase is a
mitochondrial enzyme, and fatty
acid synthesis is a cytosolic pathway, but the mitochondrial membrane is impermeable to acetyl-
CoA. Acetyl-CoA is made available in
the cytosol from citrate synthesized in the
mitochondrion, transported into the
cytosol and cleaved in a reaction catalyzed by
ATP-citrate
lyase.
Regulation of the Citric Acid Cycle Depends Primarily on a Supply of Oxidized Cofactors
In most tissues, where the primary
role of the citric acid cycle is in
energy-yielding metabolism, respiratory control via the
respiratory chain and oxidative phosphorylation regulates citric acid cycle activity. Thus, activity is immediately
dependent on the supply of NAD+, which in turn,
because of the tight coupling between oxidation and
phosphorylation, is dependent on the
availability of ADP and hence, ultich16. mately, on the rate of utilization of ATP in chemical and physical work. In addition, individual enzymes of the cycle are regulated. The most likely sites for
regulation are the nonequilibrium reactions
catalyzed by pyruvate dehydrogenase, citrate
synthase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase. The
dehydrogenases are activated by Ca2+, which increases in concentration during muscular contraction and secretion, when there is increased energy demand. In a
tissue such as brain, which is
largely dependent on carbohydrate to supply
acetyl-CoA, control of the citric acid cycle
may occur at pyruvate dehydrogenase. Several
enzymes are responsive to the energy
status, as shown by the [ATP]/[ADP] and
[NADH]/[NAD+] ratios. Thus, there
is allosteric inhibition of citrate synthase
by ATP and long-chain fatty
acyl-CoA. Allosteric activation of mitochondrial
NAD-dependent isocitrate dehydrogenase
by ADP is counteracted by ATP and NADH. The α-ketoglutarate dehydrogenase complex
is regulated in the same way as is pyruvate
dehydrogenase. Succinate dehydrogenase is inhibited by oxaloacetate, and the availability of oxaloacetate, as controlled by malate dehydrogenase, depends on the
[NADH]/[NAD+] ratio. Since the Km
for oxaloacetate of citrate synthase is of the same
order of magnitude as the intramitochondrial
concentration, it is likely that
the concentration of oxaloacetate
controls the rate of citrate formation. Which of these
mechanisms are important in vivo has
still to be resolved.
SUMMARY
• The citric acid cycle is the final
pathway for the oxidation of
carbohydrate, lipid, and protein whose common
end-metabolite, acetyl-CoA, reacts with oxaloacetate
to form citrate. By a series of
dehydrogenations and decarboxylations, citrate is
degraded, releasing reduced coenzymes and 2CO2
and regenerating oxaloacetate.
• The reduced coenzymes are oxidized
by the respiratory chain linked to formation of ATP.
Thus, the cycle is the major route for the
generation of ATP and is located in the matrix of
mitochondria adjacent to the
enzymes of the respiratory chain and oxidative phosphorylation.
• The citric acid cycle is
amphibolic, since in addition to oxidation
it is important in the provision of carbon skeletons for gluconeogenesis, fatty acid synthesis, and interconversion of amino acids.
The Pyruvate
Dehydrogenase (PDH) Complex
The bulk of ATP used by many cells to maintain homeostasis is produced
by the oxidation of pyruvate in the TCA cycle. During this oxidation process,
reduced nicotinamide adenine
dinucleotide (NADH) and reduced flavin
adenine dinucleotide (FADH2) are generated. The NADH and FADH2
are principally used to drive the processes of oxidative
phosphorylation, which are responsible for converting the reducing
potential of NADH and FADH2 to the high energy phosphate in ATP.
The fate of pyruvate depends on the cell energy charge. In cells or
tissues with a high energy charge pyruvate is directed toward gluconeogenesis,
but when the energy charge is low pyruvate is preferentially oxidized to CO2
and H2O in the TCA cycle, with generation of 15 equivalents of ATP
per pyruvate. The enzymatic activities of the TCA cycle (and of oxidative
phosphorylation) are located in the mitochondrion. When transported into the
mitochondrion, pyruvate encounters two principal metabolizing enzymes: pyruvate
carboxylase (a gluconeogenic enzyme) and pyruvate dehydrogenase (PDH), the
first enzyme of the PDH complex. With a high cell-energy charge coenzyme A
(CoA) is highly acylated, principally as acetyl-CoA, and able allosterically to
activate pyruvate carboxylase, directing pyruvate toward gluconeogenesis. When
the energy charge is low CoA is not acylated, pyruvate carboxylase is inactive,
and pyruvate is preferentially metabolized via the PDH complex and the enzymes
of the TCA cycle to CO2 and H2O. Reduced NADH and FADH2
generated during the oxidative reactions can then be used to drive ATP
synthesis via oxidative phosphorylation.
The PDH complex is comprised of multiple copies of 3 separate enzymes:
pyruvate dehydrogenase (20-30 copies), dihydrolipoyl transacetylase (60 copies)
and dihydrolipoyl dehydrogenase (6 copies). The complex also requires 5
different coenzymes: CoA, NAD+, FAD+, lipoic acid and thiamine pyrophosphate (TPP). Three of
the coenzymes of the complex are tightly bound to enzymes of the complex (TPP,
lipoic acid and FAD+) and two are employed as carriers of the
products of PDH complex activity (CoA and NAD+). The pathway of PDH
oxidation of pyruvate to acetyl-CoA is diagrammed below.
|
|
|
Flow diagram depicting the overall activity of the pyruvate dehydrogenase complex. During
the oxidation of pyruvate to CO2 by pyruvate dehydrogenase the
electrons flow from pyruvate to the lipoamide moiety of dihydrolipoyl
transacetylase then to the FAD cofactor of dihydrolipoyl dehydrogenase and
finally to reduction of NAD+ to NADH. The acetyl group is linked
to coenzyme A (CoASH) in a
high energy thioester bond. The acetyl-CoA then enters the TCA cycle for
complete oxidation to CO2 and H2O. |
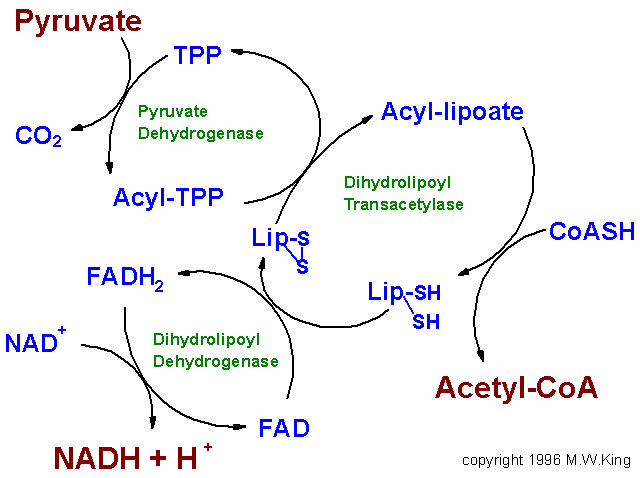
The first enzyme of the complex is PDH itself which oxidatively decarboxylates
pyruvate. During the course of the reaction the acetyl group derived from
decarboxylation of pyruvate is bound to TPP. The next reaction of the complex
is the transfer of the 2--carbon acetyl group from acetyl-TPP to lipoic acid,
the covalently bound coenzyme of lipoyl transacetylase. The transfer of the
acetyl group from acyl-lipoamide to CoA results in the formation of 2
sulfhydryl (SH) groups in lipoate requiring reoxidation to the disulfide (S-S)
form to regenerate lipoate as a competent acyl acceptor. The enzyme
dihydrolipoyl dehydrogenase, with FAD+ as a cofactor, catalyzes that
oxidation reaction. The final activity of the PDH complex is the transfer of
reducing equivalents from the FADH2 of dihydrolipoyl dehydrogenase
to NAD+. The fate of the NADH is oxidation via mitochondrial
electron transport, to produce 3 equivalents of ATP:
The net result of the reactions of the PDH complex are:
Pyruvate +
CoA + NAD+ ------> CO2 + acetyl-CoA + NADH + H+
Regulation of the PDH Complex The reactions of the PDH complex serves to
interconnect the metabolic pathways of glycolysis, gluconeogenesis and fatty
acid synthesis to the TCA cycle. As a consequence, the activity of the PDH
complex is highly regulated by a variety of allosteric effectors and by
covalent modification. The importance of the PDH complex to the maintenance of
homeostasis is evident from the fact that although diseases associated with
deficiencies of the PDH complex have been observed, affected individuals often
do not survive to maturity. Since the energy metabolism of highly aerobic
tissues such as the brain is dependent on normal conversion of pyruvate to
acetyl-CoA, aerobic tissues are most sensitive to deficiencies in components of
the PDH complex. Most genetic diseases associated with PDH complex deficiency
are due to mutations in PDH. The main pathologic result of such mutations is
moderate to severe cerebral lactic
acidosis and encephalopathies.
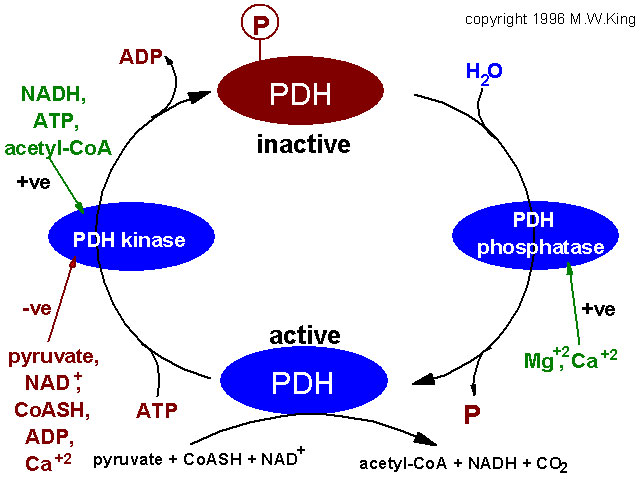
The main regulatory features of the PDH complex are diagrammed below.
|
|
|
Factors regulating the activity of pyruvate
dehydrogenase, (PDH). PDH activity is regulated by its' state of
phosphorylation, being most active in the dephosphorylated state.
Phosphorylation of PDH is catalyzed by a specific PDH kinase. The activity of
the kinase is enhanced when cellular energy charge is high which is reflected
by an increase in the level of ATP, NADH and acetyl-CoA. Conversely, an
increase in pyruvate strongly inhibits PDH kinase. Additional negative
effectors of PDH kinase are ADP, NAD+ and CoASH, the levels of
which increase when energy levels fall. The regulation of PDH phosphatase is
not completely understood but it is known that Mg2+ and Ca2+
activate the enzyme. In adipose tissue insulin increases PDH activity and in cardiac muscle PDH
activity is increased by catecholamines.
|
Two products of the complex, NADH and acetyl-CoA, are negative
allosteric effectors on PDH-a, the non-phosphorylated, active form of PDH. These
effectors reduce the affinity of the enzyme for pyruvate, thus limiting the
flow of carbon through the PDH complex. In addition, NADH and acetyl-CoA are
powerful positive effectors on PDH kinase, the enzyme that inactivates PDH by
converting it to the phosphorylated PDH-b form. Since NADH and acetyl-CoA
accumulate when the cell energy charge is high, it is not surprising that high
ATP levels also up-regulate PDH kinase activity, reinforcing down-regulation of
PDH activity in energy-rich cells. Note, however, that pyruvate is a potent
negative effector on PDH kinase, with the result that when pyruvate levels
rise, PDH-a will be favored even with high levels of NADH and acetyl-CoA.
Concentrations of pyruvate which maintain PDH in the active form (PDH-a)
are sufficiently high so that, in energy-rich cells, the allosterically
down-regulated, high Km form of PDH is nonetheless capable of
converting pyruvate to acetyl-CoA. With large amounts of pyruvate in cells
having high energy charge and high NADH, pyruvate carbon will be directed to
the 2 main storage forms of carbon (glycogen via gluconeogenesis and fat
production via fatty acid synthesis) where acetyl-CoA is the principal carbon
donor.
Although the regulation of PDH-b phosphatase is not well understood, it
is quite likely regulated to maximize pyruvate oxidation under energy-poor
conditions and to minimize PDH activity under energy-rich conditions.