Investigation of thyroid hormones in the
regulation of metabolism. Hormonal regulation of calsium and phosphorus homeostasis.
Hormones
of thyroid and parathyroid glands

Thyroid
synthesizes two kinds of hormones: iodine containing hormones and calcitonin.
Iodine containing hormones - thyroxine and triiodthyronine.
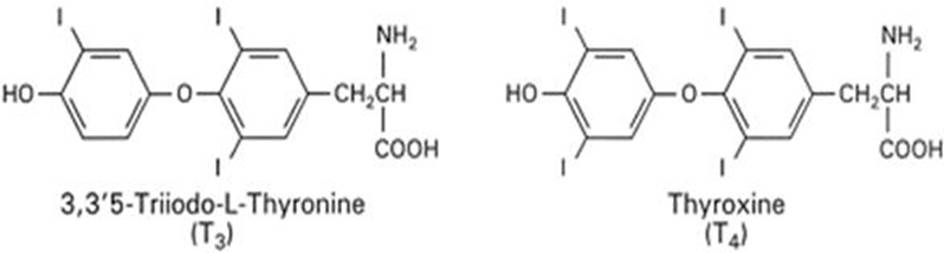
Thyroid hormone is produced
by the thyroid gland, which consists of follicles in which thyroid hormone is
synthesized through iodination of tyrosine residues in the glycoprotein thyroglobulin.
Thyroid stimulating hormone (TSH), secreted by the anterior pituitary in
response to feedback from circulating thyroid hormone, acts directly on the TSH
receptor (TSH-R) expressed on the thyroid follicular cell basolateral membrane.
TSH regulates iodide uptake mediated by the sodium/iodide symporter, followed
by a series of steps necessary for normal thyroid hormone synthesis and
secretion. Thyroid hormone is essential for normal development, growth, neural
differentiation, and metabolic regulation in mammals.
TH synthesis and secretion is exquisitely
regulated by a negative-feedback system that involves the hypothalamus,
pituitary, and thyroid gland [hypothalamic/pituitary/thyroid (HPT) axis].
Thyrotropin releasing hormone (TRH) is a tripeptide (PyroGlu-His-Pro)
synthesized in the paraventricular nucleus of the hypothalamus. It is
transported via axons to the median eminence and then to the anterior pituitary
via the portal capillary plexus. TRH binds to TRH receptors in pituitary
thyrotropes, a subpopulation of pituitary cells that secrete thyroid
stimulating hormone (TSH). TRH receptors are members of the seven-transmembrane
spanning receptor family and are coupled to Gq11. TRH stimulation
leads to release and synthesis of new TSH in thyrotropes. TSH is a 28-kDa
glycoprotein composed of α- and β-subunits
designated as glycoprotein hormone α- and TSH β-subunits.
The α-subunit
also is shared with other hormones such as luteinizing hormone, follicle
stimulating hormone, and chorionic gonadotropin. Both TRH and TSH secretion are
negatively regulated by TH. An important mechanism for the negative regulation
of TSH may be the intrapituitary conversion of circulating T4 to T3 by type II deiodinase. Additionally,
somatostatin and dopamine from the hypothalamus can negatively regulate TSH
secretion.
TSH is the primary regulator of TH release and
secretion. It also has a critical role in thyroid growth and development. TSH
binds to the TSH receptor (TSHr), which also is a seven-transmembrane spanning
receptor coupled to Gs. Activation of TSHr by TSH or autoantibodies
in Graves' disease leads to an increase in intracellular cAMP and stimulation
of protein kinase A-mediated pathways. A number of thyroid genes, including Na+/I− symporter (NIS), thyroglobulin (Tg),
and thyroid peroxidase (TPO), are stimulated by TSH and promote the synthesis
of TH.
The THs, T4 and the more potent T3, are
synthesized in the thyroid gland. Iodide is actively transported and
concentrated into the thyroid by NIS
(102,475). The
trapped iodide is oxidized by TPO in the presence of hydrogen peroxide and
incorporated into the tyrosine residues of a 660-kDa glycoprotein, Tg. This
iodination of specific tyrosines located on Tg yields monoiodinated and
diiodinated residues (MIT, monoiodo-tyrosines; DIT, diiodo-tyrosines) that are
enzymatically coupled to form T4 and
T3. The iodinated Tg containing MIT, DIT, T4, and T3,
then is stored as an extracellular storage polypeptide in the colloid within
the lumen of thyroid follicular cells. Genetic defects along the synthetic
pathway of THs have been described in humans and are major causes of congenital
hypothyroidism in iodine-replete environments.
The secretion of THs requires endocytosis of the
stored iodinated Tg from the apical surface of the thyroid follicular cell. The
internalized Tg is incorporated in phagolysosomes and undergoes proteolytic
digestion, recapture of MIT and DIT, and release of T4 and T3 into the circulation via the basal
surface. The majority of released TH is in the form of T4, as total
serum T4 is 40-fold
higher than serum T3 (90
vs. 2 nM). Only 0.03% of the total serum T4 is free (unbound), with the remainder
bound to carrier proteins such as thyroxine binding globulin (TBG), albumin,
and thyroid binding prealbumin. Approximately 0.3% of the total serum T3 is free, with the remainder bound to
TBG and albumin. It is the free TH that enters target cells and generates a
biological response.
The major pathway for the production of T3 is via 5′-deiodination of the
outer ring of T4 by
deiodinases and accounts for the majority of the circulating T3.
Type I deioidinase is found in peripheral tissues such as liver and kidney and
is responsible for the conversion of the majority of T4 to T3 in circulation. Type II deiodinase is
found in brain, pituitary, and brown adipose tissue and primarily converts T4 to T3for intracellular use.
These deiodinases recently have been cloned and demonstrated to be
selenoproteins. 5′-Deiodination by type I deiodinase and type III
deioidinase, which is found primarily in placenta, brain, and skin, leads to
the generation of rT3, the key step in the inactivation of TH. rT3 and T3 can be further deiodinated in the
liver and are sulfo- and glucuronide-conjugated before excretion in the bile.
There also is an enterohepatic circulation of TH as intestinal flora
deconjugates some of these compounds and promotes the reuptake of TH.
Although THs may exert their effects on a number
of intracellular loci, their primary effect is on the transcriptional
regulation of target genes. Early studies showed that the effects of THs at the
genomic level are mediated by nuclear TRs, which are intimately associated with
chromatin and bind TH with high affinity and specificity. Similar to steroid
hormones that also bind to nuclear receptors, TH enters the cell and proceeds
to the nucleus. It then binds to TRs, which may already be prebound to TREs
located in promoter regions of target genes. The formation of ligand-bound TR
complexes that are also bound to TREs is the critical first step in the
positive or negative regulation of target genes and the subsequent regulation
of protein synthesis. Given their abilities to bind both ligand and DNA as well
as their ability to regulate transcription, TRs can be regarded as
ligand-regulatable transcription factors.

General model
for thyroid hormone action in the nucleus. TR, thyroid hormone receptor; RXR,
retinoid X receptor
Thyroid Hormone Receptors and Mechanism of Action
Thyroid hormone regulates a wide range of genes after its activation from
the prohormone, thyroxine (T4), to the active form, triiodothyronine (T3). The
signaling pathway is complex and highly regulated due to the expression of cell
and tissue-specific thyroid hormone transporters, multiple thyroid hormone
receptor (TR) isoforms, and interactions with corepressors and coactivators.
Furthermore, in many cases, thyroid signals are involved in cross-talk with a
range of other signaling pathways.
Receptors for thyroid hormones are intracellular DNA-binding proteins that
function as hormone-responsive transcription factors, very similar conceptually
to the receptors for steroid hormones.
Thyroid hormones enter cells through membrane transporter proteins. A
number of plasma membrane transporters have been identified, some of which
require ATP hydrolysis; the relative importance of different carrier systems is
not yet clear and may differ among tissues. Once inside the nucleus, the
hormone binds its receptor, and the hormone-receptor complex interacts with
specific sequences of DNA in the promoters of responsive genes. The effect
of the hormone-receptor complex binding to DNA is to modulate gene expression,
either by stimulating or inhibiting transcription of specific genes.

Nuclear action of thyroid
hormone.
For the purpose of illustration,
consider one mechanism by which thyroid hormones increase the strength of
contraction of the heart. Cardiac contractility depends, in part, on the
relative ratio of different types of myosin proteins in cardiac muscle.
Transcription of some myosin genes is stimulated by thyroid hormones, while
transcription of others in inhibited. The net effect is to alter the ratio
toward increased contractility.
Metabolism: Thyroid hormones
stimulate diverse metabolic activities most tissues, leading to an increase in
basal metabolic rate. One consequence of this activity is to increase body heat
production, which seems to result, at least in part, from increased oxygen
consumption and rates of ATP hydrolysis. By way of analogy, the action of
thyroid hormones is akin to blowing on a smouldering fire. A few examples of
specific metabolic effects of thyroid hormones include:
Lipid metabolism: Increased
thyroid hormone levels stimulate fat mobilization, leading to increased
concentrations of fatty acids in plasma. They also enhance oxidation of fatty
acids in many tissues. Finally, plasma concentrations of cholesterol and
triglycerides are inversely correlated with thyroid hormone levels - one
diagnostic indiction of hypothyroidism is increased blood cholesterol
concentration.
Carbohydrate metabolism: Thyroid hormones stimulate almost all aspects of carbohydrate
metabolism, including enhancement of insulin-dependent entry of glucose into
cells and increased gluconeogenesis and glycogenolysis to generate free
glucose.
Protein metabolism: in normal concentration stimulate the synthesis of proteins and nucleic
acids; in excessive concentration activate the catabolic processes.
Growth: Thyroid
hormones are clearly necessary for normal growth in children and young animals,
as evidenced by the growth-retardation observed in thyroid deficiency. Not
surprisingly, the growth-promoting effect of thyroid hormones is intimately
intertwined with that of growth hormone, a clear indiction that complex physiologic processes like growth depend
upon multiple endocrine controls.
Development: Of critical
importance in mammals is the fact that normal levels of thyroid hormone
are essential to the development of the fetal and neonatal brain.
Other Effects: As
mentioned above, there do not seem to be organs and tissues that are not
affected by thyroid hormones. A few additional, well-documented effects of
thyroid hormones include:
Cardiovascular system: Thyroid hormones increases heart rate, cardiac contractility and cardiac
output. They also promote vasodilation, which leads to enhanced blood flow to
many organs.
Central nervous system: Both decreased and increased concentrations of thyroid hormones lead to
alterations in mental state. Too little thyroid hormone, and the individual
tends to feel mentally sluggish, while too much induces anxiety and
nervousness.
Reproductive system: Normal reproductive behavior and
physiology is dependent on having essentially normal levels of thyroid hormone.
Hypothyroidism in particular is commonly associated with infertility.
Thyroid Disease States
Disease is associated with both
inadequate production and overproduction of thyroid hormones. Both types of
disease are relatively common afflictions of man and animals.
Hypothyroidism is the
result from any condition that results in thyroid hormone deficiency. Two
well-known examples include:
Iodine deficiency: Iodide is
absolutely necessary for production of thyroid hormones; without adequate
iodine intake, thyroid hormones cannot be synthesized. Historically, this
problem was seen particularly in areas with iodine-deficient soils, and frank
iodine deficiency has been virtually eliminated by iodine supplementation of
salt.
Primary thyroid disease:
Inflammatory diseases of the thyroid that destroy parts of the gland are
clearly an important cause of hypothyroidism.
Common symptoms of hypothyroidism arising after early
childhood include lethargy, fatigue, cold-intolerance, weakness, hair loss and
reproductive failure. If these signs are severe, the clinical condition is
called myxedema. In the case of iodide deficiency, the thyroid
becomes inordinantly large and is called a goiter.


About 95
percent of the active thyroid hormone is thyroxine, and most of the remaining 5
percent is triiodothyronine. Both of these require iodine for their synthesis.
Thyroid hormone secretion is regulated by a negative feedback mechanism that
involves the amount of circulating hormone, hypothalamus, and adenohypophysis.
If there is
an iodine deficiency, the thyroid cannot make sufficient hormone. This
stimulates the anterior pituitary to secrete thyroid-stimulating hormone, which
causes the thyroid gland to increase in size in a vain attempt to produce more
hormones. But it cannot produce more hormones because it does not have the
necessary raw material, iodine. This type of thyroid enlargement is called
simple goiter or iodine
deficiency goiter.


Calcitonin is
secreted by the parafollicular cells of the thyroid gland. This hormone opposes
the action of the parathyroid glands by reducing the calcium level in the
blood. If blood calcium becomes too high, calcitonin is secreted until calcium
ion levels decrease to normal.
The most severe and devestating form
of hypothyroidism is seen in young children with congenital thyroid deficiency. If that
condition is not corrected by supplemental therapy soon after birth, the child
will suffer from cretinism, a form of
irreversible growth and mental retardation.

Congenital hypothyroidism can be
endemic, genetic, or sporadic. If untreated, it results in mild to severe
impairment of both physical and mental growth and development. Poor
length growth is apparent as early as the first year of life. Adult stature
without treatment ranges from 1 to 1.6 metres (3'4 to 5'3), depending on severity,
sex and other genetic factors. Bone maturation and puberty are severely
delayed. Ovulation is impeded
and infertility is common. Neurological
impairment may be mild, with reduced muscle tone and coordination, or so severe
that the person cannot stand or walk. Cognitive impairment may also range from
mild to so severe that the person is nonverbal and dependent on others for
basic care. Thought and reflexes are slower. Other
signs may include thickened skin, enlarged tongue, or a protruding abdomen.
Sporadic and genetic cretinism
results from abnormal development or function of the foetal thyroid gland. This
type of cretinism has been almost completely eliminated in developed countries
by early diagnosis by newborn screening schemes
followed by lifelong treatment with thyroxine (T4).
Thyroxine must be dosed
as tablets only, even to newborns, as the liquid oral suspensions and
compounded forms cannot be depended on for reliable dosing. In the case of
dosing infants, the T4 tablets are generally crushed and mixed with breast
milk, formula milk or water. If the medication is mixed with formulas
containing iron or soya products, larger doses may be required, as these
substances may alter the absorption of thyroid hormone from the gut. Frequent monitoring (every 2–3 weeks
during the first months of life) is recommended to ensure that infants with
congenital hypothyroidism remain within the high end of normal range, or euthyroid.
Cretinism arises
from a diet deficient in iodine. It has affected many people worldwide and continues to be a major public health problem in many countries. Iodine is
an essential trace element, necessary primarily for the synthesis of thyroid
hormones. Iodine deficiency is the most common preventable cause of brain
damage worldwide. Although iodine is found in many
foods, it is not universally present in all soils in adequate amounts. Most
iodine, in iodide form, is in the oceans where the iodide ions oxidize to
elemental iodine, which then enters the atmosphere and falls to earth as rain,
introducing iodine to soils. Earth deficient in iodine is most common inland
and in mountainous areas and areas of frequent flooding, but can also occur in
coastal regions owing to past glaciation, and leaching by snow, water and heavy
rainfall, which removes iodine from the soil.[8] Plants
and animals grown in iodine deficient soils are correspondingly deficient.
Populations living in those areas without outside food sources are most at risk
ofiodine deficiency
diseases.
Most cases of
hypothyroidism are readily treated by oral administration of synthetic thyroid
hormone. In times past, consumption of dessicated animal thyroid gland was used
for the same purpose.

Hyperthyroidism results from secretion of thyroid hormones. In most species, this condition
is less common than hypothyroidism. In humans the most common form of
hyperthyroidism is Graves
disease, an immune disease in which autoantibodies bind to and activate the
thyroid-stimulating hormone receptor, leading to continual stimulation of thyroid
hormone synthesis. Another interesting, but rare cause of hyperthyroidism is
so-called hamburger thyrotoxicosis.

exophthalmic
goiter

Common signs
of hyperthyroidism are basically the opposite of those seen in hypothyroidism,
and include nervousness, insomnia, high heart rate, eye disease and anxiety.
Graves disease is commonly treated with anti-thyroid drugs (e.g.
propylthiourea, methimazole), which suppress synthesis of thyroid hormones
primarily by interfering with iodination of thyroglobulin by thyroid peroxidase.
Calcitonin.
Calcitonin is
synthesized by the parafollicle cells of thyroid.
Chemical
structure: peptide.
Functions: -
promotes the transition of calcium from blood in bones;
-
inhibits
the reabsorption of phosphorus in kidneys.
Thus,
calcitonin decreases the Ca and P contents in blood.

Parathyroid
glands.

Parathyroid
Gland
Four small
masses of epithelial tissue are embedded in the connective tissue capsule on
the posterior surface of the thyroid glands. These are parathyroid glands, and
they secrete parathyroid hormone or parathormone. Parathyroid hormone is the
most important regulator of blood calcium levels. The hormone is secreted in
response to low blood calcium levels, and its effect is to increase those
levels.
Parathyroid hormone. Chemical structure: protein.
Functions:
1.
promotes
the transition of calcium from bones to blood;
2.
promotes
the absorption of Ca in the intestine;
3.
inhibits
the reabsorption of phosphorus in kidneys.
Thus,
parathyroid hormone increases the Ca amount in blood and decreases the P amount in blood.
Body Distribution of Calcium and Phosphate
There are
three major pools of calcium in the body:
Intracellular
calcium:
·
A large majority of calcium within cells is
sequestered in mitochondria and endoplasmic reticulum. Intracellular free
calcium concentrations fluctuate greatly, from roughly 100 nM to greater than 1
uM, due to release from cellular stores or influx from extracellular fluid.
These fluctuations are integral to calcium's role in intracellular signaling,
enzyme activation and muscle contractions.
Calcium in
blood and extracellular fluid:
Roughly half
of the calcium in blood is bound to proteins. The concentration of ionized
calcium in this compartment is normally almost invariant at approximately 1 mM,
or 10,000 times the basal concentration of free calcium within cells. Also, the
concentration of phosphorus in blood is essentially identical to that of
calcium.

Bone calcium:

A vast
majority of body calcium is in bone. Within bone, 99% of the calcium is tied up
in the mineral phase, but the remaining 1% is in a pool that can rapidly
exchange with extracellular calcium.
As with
calcium, the majority of body phosphate (approximately 85%) is present in the
mineral phase of bone. The remainder of body phosphate is present in a variety
of inorganic and organic compounds distributed within both intracellular and
extracellular compartments. Normal blood concentrations of phosphate are very
similar to calcium.
Fluxes of
Calcium and Phosphate
Maintaining
constant concentrations of calcium in blood requires frequent adjustments, which
can be described as fluxes of calcium between blood and other body
compartments. Three organs participate in supplying calcium to blood and
removing it from blood when necessary:
·
The small intestine is the site where dietary
calcium is absorbed. Importantly, efficient absorption of calcium in the small
intestine is dependent on expression of a calcium-binding protein in epithelial
cells.
·
Bone serves as a vast
reservoir of calcium. Stimulating net resorption of bone mineral releases
calcium and phosphate into blood, and suppressing this effect allows calcium to
be deposited in bone.
The kidney
is critcally important in calcium homeostasis. Under normal blood calcium
concentrations, almost all of the calcium that enters glomerular filtrate is
reabsorbed from the tubular system back into blood, which preserves blood
calcium levels. If tubular reabsorption of calcium decreases, calcium is lost
by excretion into urine.
Hormonal
Control Systems
Maintaining
normal blood calcium and phosphorus concentrations is managed through the
concerted action of three hormones that control fluxes of calcium in and out of
blood and extracellular fluid:
Calcitonin is a hormone that functions to reduce blood
calcium levels. It is secreted in response to hypercalcemia and has at least
two effects:
·
Suppression of renal tubular reabsorption of calcium.
In other words, calcitonin enhances excretion of calcium into urine.
·
Inhibition of bone resorption, which would minimize
fluxes of calcium from bone into blood.
Although
calcitonin has significant calcium-lowing effects in some species, it appears
to have a minimal influence on blood calcium levels in humans.
Vitamin D acts also to increase blood concentrations of calcium. It is generated through the activity
of parathyroid hormone within the kidney. Far and away the most important
effect of vitamin D is to facilitate absorption of calcium from the small
intestine. In concert with parathyroid hormone, vitamin D also enhances fluxes
of calcium out of bone.
http://www.youtube.com/watch?v=JwPVibQ6_3Y&feature=related
http://www.youtube.com/watch?v=n7vybcT9_F4
Parathyroid hormone serves to increase blood
concentrations of calcium. Mechanistically, parathyroid hormone preserves blood calcium by several
major effects:
·
Stimulates production of the biologically-active form
of vitamin D within the kidney.
·
Facilitates mobilization of calcium and phosphate
from bone. To prevent detrimental increases in phosphate, parathyroid hormone
also has a potent effect on the kidney to eliminate phosphate (phosphaturic
effect).
·
Maximizes tubular reabsorption of calcium within the
kidney. This activity results in minimal losses of calcium in urine.
Hypoparathyroidism, or insufficient secretion of parathyroid hormone, leads to increased
nerve excitability. The low blood calcium levels trigger spontaneous and
continuous nerve impulses, which then stimulate muscle contraction.
Since parathyroid
gland disease (hyperparathyroidism) was first described in 1925, the symptoms
have become known as "moans, groans, stones, and bones...with psychic
overtones". Although about 5-7% of people with
parathyroid disease (hyperparathyroidism) claim they don't have symptoms and to
feel fine when the diagnosis of hyperparathyroidism is made, almost 100% of
parathyroid patients will actually say they feel better after the parathyroid
problem has been cured--proving they had symptoms. The bottom line: Nearly ALL patients with parathyroid problems have symptoms. Sometimes the symptoms
are real obvious, like kidney stones, frequent headaches, and depression.
Sometimes the symptoms are not so obvious, like high blood pressure and the
inability to concentrate. If you have symptoms, you are almost guaranteed to
feel remarkably better once the parathyroid tumor has been removed. As we often
tell our parathyroid patients: "you will be amazed at how a 16 minute
mini-procedure will change your life!"

Hormones of
adrenal cortex
Adrenal
glands consist of two parts: external - cortex, internal - medulla.
Each part
secrets specific hormones.
Hormones
synthesized in adrenal cortex are named corticosteroids.

Mechanism of
steroid hormones action (permeating into the cells):
http://www.youtube.com/watch?v=oOj04WsU9ko
In difference to hormones of protein and peptide nature,
receptors for steroid hormones are located within the cells - in the cytoplasm.
From cytoplasm the hormone-receptor complexes is translocated into the nucleus where they interact with
DNA of nuclear chromatin causing the activation of genes for respective enzyme
proteins. So, if hormones of the first group cause the activation of existing
enzyme molecules, the acting on the target cells of steroids and thyroid
hormones results in the biosynthesis of new enzyme molecules.
http://www.youtube.com/watch?v=0ss8YIoKw0g
Receptors for steroid and thyroid
hormones are located inside target cells, in the cytoplasm or nucleus, and
function as ligand-dependent
transcription factors. That is to say, the hormone-receptor complex binds
to promoter regions of responsive genes and stimulate or sometimes inhibit
transcription from those genes.

Thus, the mechanism of action of
steroid hormones is to modulate gene expression in target cells. By selectively
affecting transcription from a battery of genes, the concentration of those
respective proteins are altered, which clearly can change the phenotype of the
cell.
Steroid and thyroid hormone receptors
are members of a large group ("superfamily") of transcription
factors. In some cases, multiple forms of a given receptor are expressed in
cells, adding to the complexity of the response. All of these receptors are
composed of a single polypeptide chain that has, in the simplist analysis,
three distinct domains:

In addition to these three core
domains, two other important regions of the receptor protein are a nuclear
localization sequence, which targets the the protein to nucleus, and a
dimerization domain, which is responsible for latching two receptors together
in a form capable of binding DNA.
Being lipids, steroid hormones enter
the cell by simple diffusion across the plasma membrane. Thyroid hormones enter
the cell by facilitated diffusion. The receptors exist either in the cytoplasm
or nucleus, which is where they meet the hormone. When hormone binds to
receptor, a characteristic series of events occurs:

As might be
expected, there are a number of variations on the themes described above,
depending on the specific receptor in question. For example, in the absense of
hormone, some intracellular receptors do bind their hormone response elements
loosely and silence transcription, but, when complexed to hormone, become
activated and strongly stimulate transcription. Some receptors bind DNA not
with another of their kind, but with different intracellular receptor.
Corticosteroids have potent regulatory effect
on all kinds of metabolism. Cholesterol is the precursor of corticosteroids.
According to the biological effect corticosteroids are divided on two groups: glucocorticoids and mineralocorticoids. Glucocorticoids regulate the protein,
lipid and carbohydrate metabolism, mineralocorticoids - metabolism of water and
mineral salt.
The most important glucocorticoids: corticosterone,
hydrocortisone, cortisol. The
most important mineralocorticoid: aldosterone.
All biological active hormones of adrenal
cortex consist of 21 carbon atom and can be reviewed as derivatives of
carbohydrate pregnane.
The synthesis of corticosteroids is
regulated by ACTH.
In the blood corticosteroids are
connected with proteins and transported to different organs.
Time half-life for corticosteroids is
about 1 hour.

These forms
of hormones are lipids. They can enter the cell membrane quite easily and enter
right into the nuclei. Steroid hormones are generally carried in the blood
bound to specific carrier proteins such
as sex hormone binding globulin or corticosteroid binding globulin. Further
conversions and catabolism occurs in the liver, other "peripheral"
tissues, and in the target tissues.
Ways of
metabolism of corticosteroids:
1.
Reduction.
Corticosteroids accept 4 or 6 hydrogen atoms and form couple compounds with
glucuronic acid. These compounds ere excreted by kidneys.
2.
Oxidation
of 21-st carbon atom.
3.
Reduction
of ring and decomposition of side chain. As result 17-ketosteroids are
formed that are excreted with urine. The
determination of 17-ketosteroids in urine - important diagnostic indicator.
This is the indicator of adrenal cortex function. In men 17-ketosteroids are
also the terminal products of sex hormones metabolism giving important
information about testicles function.
4.
Corticosteroids can be excreted by kidneys in
native structure.
Synthesis of
steroid hormons

Glucocorticoids
The name
"glucocorticoid" derives from early observations that these hormones
were involved in glucose metabolism. In the fasted state, cortisol stimulates several processes that
collectively serve to increase and maintain normal concentrations of glucose in
blood.
Metabolic
effects:
Excessive
glucocorticoid levels resulting from administration as a drug or
hyperadrenocorticism have effects on many systems. Some examples include
inhibition of bone formation, suppression of calcium absorption (both of which
can lead to osteoporosis), delayed
wound healing, muscle weakness, and increased risk of infection. These
observations suggest a multitude of less-dramatic physiologic roles for
glucocorticoids.


http://www.youtube.com/watch?v=0ss8YIoKw0g
The effect of glucocorticoids on protein metabolism:
1.
stimulate
the catabolic processes (protein decomposition) in connective, lymphoid and
muscle tissues and activate the processes of protein synthesis in liver;
2.
stimulate
the activity of aminotransferases;
3.
activate
the synthesis of urea.
The effect of glucocorticoids on
carbohydrate metabolism:
1.
activate
the gluconeogenesis;
2.
inhibit
the activity of hexokinase;
3.
activate
the glycogen synthesis in liver.
Glucocorticoids causes the
hyperglycemia.

The effect of glucocorticoids on
lipid metabolism:
1.
promote
the absorption of lipids in intestine;
2.
activate
lipolisis;
3.
activate the conversion of fatty
acids in carbohydrates.
Hyperfunction of adrenal cortex
causes Icenko-Kushing syndrome. This
state is called steroid diabetes. Symptoms:
hyperglycemia, glucosuria, hypercholesterolemia, hypernatriemia,
hyperchloremia, hypokaliemia.
http://www.youtube.com/watch?v=ku-QJyQ0j7M&feature=related

Hypercholesterolemia
Adrenal
cortex hormones and their artificial analogs are often used in clinic: for
treatment of allergic and autoimmune diseases, in hard shock states.
Blood and
urine cortisol, together with the determination of adrenocorticotropic hormone
(ACTH), are the three most important tests in the investigation of Cushing's
syndrome (caused by an overproduction of cortisol) and Addison's disease
(caused by the underproduction of cortisol).
Cushing's
syndrome

Reference
ranges for cortisol vary from laboratory to laboratory but are usually within
the following ranges for blood:
·
adults (8 A.M.): 6-28 mg/dL; adults (4 P.M.): 2-12
mg/dL
·
child one to six years (8 A.M.): 3-21 mg/dL; child
one to six years (4 P.M.): 3-10 mg/dL
·
newborn: 1/24 mg/dL.
Reference
ranges for cortisol vary from laboratory to laboratory, but are usually within
the following ranges for 24-hour urine collection:
·
adult: 10-100 mg/24 hours
·
adolescent: 5-55 mg/24 hours
·
Child: 2-27
mg/24 hours.
Abnormal
results
Increased
levels of cortisol are found in Cushing's syndrome, excess thyroid (hyperthyroidism),
obesity, ACTH-producing tumors, and high levels of stress.

Decreased
levels of cortisol are found in Addison's disease, conditions of low thyroid,
and hypopituitarism, in which pituitary activity is diminished.
Cushing's
syndrome
A hormonal
disorder caused by an abnormally high level of corticosteroid hormones. Symptoms
include high blood sugar levels, a moon face, weight gain, and increased blood
pressure
In 1932, a
physician by the name of Harvey Cushing described eight patients with central
body obesity, glucose intolerance, hypertension, excess hair growth,
osteoporosis, kidney stones, menstrual irregularity, and emotional liability.
It is now known that these symptoms are the result of excess production of
cortisol by the adrenal glands. Cortisol is a powerful steroid hormone, and
excess cortisol has detrimental effects on many cells throughout the body.
Although some of these symptoms are common by themselves, the combination of
these suggests that a workup for this disease may be in order. Keep in mind
that Cushings syndrome is rare, occurring in only about 10 patients per one
million population. On the other hand, simple obesity can be associated with
some of these symptoms in the absence of an adrenal tumor--this is related to the
slightly different mechanism by which normally produced steroids are
metabolized by individuals who are obese.
Since
cortisol production by the adrenal glands is normally under the control of the
pituitary (like the thyroid gland), overproduction
can be caused by a tumor in the pituitary or within the adrenal glands
themselves. When a pituitary tumor secretes too much ACTH
(Adrenal Cortical Tropic Hormone), it simply causes the otherwise normal
adrenal glands to produce too much cortisol. This type of Cushings syndrome is
termed "Cushings Disease" and it is diagnosed like other endocrine
disorders by measuring the appropriateness of hormone production. In this case,
serum cortisol will be elevated, and, serum ACTH will be elevated at the same time. When the adrenal glands develop a tumor, like
any other endocrine gland, they usually produce excess amounts of the hormone
normally produced by these cells. If the adrenal tumor is composed of cortisol
producing cells, excess cortisol will be produced which can be measured in the
blood. Under these conditions, the normal pituitary will sense the excess
cortisol and will stop making ACTH in an attempt to slow the adrenal down. In
this manner, physicians can readily distinguish whether excess cortisol is the
result of a pituitary tumor, or an adrenal tumor.
Even more
rare (but placed here for completion sake) is when excess ACTH is produced
somewhere other than the pituitary. This is extremely uncommon, but certain
lung cancers can make ACTH (we don't know why) and the patients develop
Cushings Syndrome in the same way they do as if the ACTH was coming from the
pituitary.
Causes of
Cushings Syndrome
ACTH
Dependent (80%)
 Pituitary
Tumors (60%)
Pituitary
Tumors (60%)

Lung Cancers (5%)
ACTH
Independent (20%)
 Benign
Adrenal Tumors (adenoma) (25%)
Benign
Adrenal Tumors (adenoma) (25%)
Malignant
Adrenal Tumors (adrenal cell carcinoma) (10%)
Testing for
Cushings Syndrome
 The most sensitive test to check for
the possibility of this disease is to measure the amount of cortisol
The most sensitive test to check for
the possibility of this disease is to measure the amount of cortisol

excreted in
the during during a 24 hour time period. Cortisol is normally secreted in
different amounts during the day and night, so this test usually will be
repeated once or twice to eliminate the variability which is normally seen.
This normal variability is why simply checking the amount of cortisol in the
blood is not a very reliable test. A 24 hour free cortisol level greater than
100 ug is diagnostic of Cushings syndrome. The second test which helps confirms
this diagnosis is the suppression test which measures the cortisol secretion
following the administration of a powerful synthetic steroid which will shut
down steroid production in everybody with a normal adrenal gland. Subsequent
tests will distinguish whether the disease is due to an ACTH dependent or
independent cause.
Invariably, once the diagnosis is
made, patients will undergo a CT scan (or possibly an MRI or Ultrasound) of the
adrenal glands to look for tumors in one or both of them (more information on adrenal x-ray tests on another page). If the laboratory test suggest a
pituitary origin, a CT or MRI of the brain (and possibly of the chest as well)
will be performed.
Treatment of Cushings Syndrome

 Obviously, the treatment of this disease depends upon the
cause. Pituitary tumors are usually removed surgically and often treated with
radiation therapy. Neurosurgeons and some ENT surgeons specialize in these
tumors. If the cause is determined to be within a single adrenal gland, this is
treated by surgical removal. If the tumor has characteristics of cancer on any
of the x-ray tests, then a larger, conventional operation is in order. If a
single adrenal gland possesses a small, well defined tumor, it can usually be
removed by the new technique of laparoscopic adrenalectomy.
Obviously, the treatment of this disease depends upon the
cause. Pituitary tumors are usually removed surgically and often treated with
radiation therapy. Neurosurgeons and some ENT surgeons specialize in these
tumors. If the cause is determined to be within a single adrenal gland, this is
treated by surgical removal. If the tumor has characteristics of cancer on any
of the x-ray tests, then a larger, conventional operation is in order. If a
single adrenal gland possesses a small, well defined tumor, it can usually be
removed by the new technique of laparoscopic adrenalectomy.
 Functions of
mineralocorticoids.
Functions of
mineralocorticoids.
Secretion of mineralocorticoids is
regulated by renin-angiotensine system
-
activates
the reabsorption of Na+, Cl- and water in kidney
canaliculuses;
-
promote
the excretion of K+ by kidneys, skin and saliva.
Deficiency of corticosteroids causes Addison's disease.


For this disease the
hyperpigmentation is typical because the deficiency of corticosteroids results
in the excessive synthesis of ACTH.
http://www.youtube.com/watch?v=FK1pPqWMXjM
Addison's disease
A rare disorder in which symptoms are
caused by a deficiency of hydrocortisone (cortisol) and aldosterone, two
corticosteroid hormones normally produced by a part of the adrenal glands
called the adrenal cortex. Symptoms include weakness, tiredness, vague
abdominal pain, weight loss, skin pigmentation and low blood pressure.
. Mineralocorticoids
Primary
aldosteronism

Conn's
syndrome is an aldosterone-producing adenoma. Conn's syndrome is named after Jerome W. Conn (1907–1994), the Americanendocrinologist who first described the
condition at the University of Michigan in 1955.
Primary hyperaldosteronism has many causes, including adrenal hyperplasia and adrenal carcinoma.[2]
The syndrome
is due to:
·
Bilateral
(micronodular) adrenal hyperplasia, 60%
·
Adrenal
(Conn's) adenoma, 40%
·
Glucocorticoid-remediable
hyperaldosteronism (dexamethasone-suppressible
hyperaldosteronism), <1%
·
rare forms, including disorders of the renin-angiotensin system, <1%
Aldosterone enhances exchange of sodium for potassium
in the kidney, so increased aldosteronism will lead to hypernatremia (elevated sodium level) and hypokalemia (low blood potassium). Once the
potassium has been significantly reduced by aldosterone, a sodium/hydrogen pump
in the nephron becomes more active, leading to
increased excretion of hydrogen ions and further exacerbating the elevated
sodium level resulting in a further increase in hypernatremia. The
hydrogen ions exchanged for sodium are generated by carbonic anhydrase in the renal tubule epithelium,
causing increased production of bicarbonate. The
increased bicarbonate and the excreted hydrogen combine to generate a metabolic alkalosis.
The high pH of the blood makes calcium less available to the tissues and
causes symptoms of hypocalcemia (low calcium levels).
The sodium
retention leads to plasma volume expansion and elevated blood pressure. The
increased blood pressure will lead to an increased glomerular filtration rate and cause a decrease inrenin release from the granular cells of
the juxtaglomerular apparatus in the kidney. If a patient is
thought to suffer from primary hyperaldosteronism, the aldosterone:renin
activity ratio is used to assess this. The decreased renin levels and in turn
the reactive down-regulation of angiotensin II are thought to be unable to
down-regulate the constitutively formed aldosterone, thus leading to an
elevated [plasma aldosterone:plasma renin activity]
ratio (lending the assay to be a clinical tool for diagnostic purposes).
Aside from
hypertension, other manifesting problems include myalgias, weakness, and
chronic headaches. The muscle cramps are due to neuron hyperexcitability
seen in the setting of hypocalcemia, muscle weakness secondary to hypoexcitability of skeletal
muscles in the setting of low blood potassium (hypokalemia), and headaches which are thought to be due to both
electrolyte imbalance (hypokalemia) and
hypertension.
Secondary
hyperaldosteronism is often related to decreased cardiac output, which is
associated with elevated renin levels.
Measuring
aldosterone alone is not considered adequate to diagnose primary
hyperaldosteronism. The screening test of choice for diagnosis is the plasma
aldosterone:plasma renin activity ratio. Renin activity, not simply plasma
renin level, is assayed. Both renin and aldosterone are measured, and a ratio greater
than 30 is indicative of primary hyperaldosteronism.
In the absence of proper treatment,
individuals with hyperaldosteronism often suffer from poorly controlled high
blood pressure, which may be associated with increased rates of stroke, heart
disease, and kidney failure. With appropriate treatment, the prognosis is
excellent.
Sex hormones.
Sex hormones are synthesized in testes,
ovaries. Smaller amount of sex hormones are produced in adrenal cortex and
placenta. Small amount of male sex hormones are produced in ovaries and female
sex hormones - in testes.
Male sex hormones are called androgens and female - estrogens.
Chemical structure - steroids.

Synthesis and secretion of the sex
hormones are controlled by the pituitary honadotropic hormones. Sex hormones
act by means of the activation of gene apparatus of cells. Catabolism of sex
hormones takes place in liver. The time half-life is 70-90 min.
The main estrogens: estradiol,
estrole, estriole (are produced by follicles) and progesterone (is produced
by yellow body and placenta). The main biological role of estrogens -
conditioning for the reproductive female function (possibility of ovum
fertilization). Estradiol results in the proliferation of endometrium and
progesterone stimulates the conversion of endometrium in decidual tissue which
is ready for ovum implantation. Estrogens also cause the development of
secondary sexual features.
Estrogens


Estrogens originate in the adrenal cortex
and gonads and primarily affect maturation and function of secondary sex organs
(female sexual determination).
Estrogens, in females, are produced primarily by the ovaries, and during
pregnancy, the placenta. Follicle-stimulating hormone(FSH)
stimulates the ovarian production of estrogens by the granulosa cells of the ovarian follicles and corpora lutea. Some
estrogens are also produced in smaller amounts by other tissues such as the liver, adrenal glands, and the breasts. These
secondary sources of estrogens are especially important in postmenopausal
women.Fat cells produce estrogen as
well.
In females, synthesis of estrogens starts in theca interna cells in the ovary, by the synthesis
of androstenedionefrom cholesterol.
Androstenedione is a substance of weak androgenic activity which serves
predominantly as aprecursor for more potent androgens such
as testosterone as well as estrogen. This compound crosses thebasal membrane into the surrounding granulosa cells,
where it is converted either immediately into estrone, or into testosterone and
then estradiol in an additional step. The conversion of androstenedione to
testosterone is catalyzed by 17β-hydroxysteroid dehydrogenase (17β-HSD), whereas the conversion of androstenedione and
testosterone into estrone and estradiol, respectively
is catalyzed by aromatase, enzymes which are both expressed in granulosa cells.
In contrast, granulosa cells lack 17α-hydroxylase and 17,20-lyase, whereas
theca cells express these enzymes and 17β-HSD but lack
aromatase. Hence, both granulosa and theca cells are essential for the
production of estrogen in the ovaries.
Estrogen levels vary through the menstrual cycle, with levels
highest near the end of the follicular phase just before ovulation.

The actions
of estrogen are mediated by the estrogen receptor (ER), a dimeric nuclear protein that
binds to DNA and controls gene expression. Like other steroid hormones,
estrogen enters passively into the cell where it binds to and activates the
estrogen receptor. The estrogen:ER complex binds to
specific DNA sequences called a hormone response element to activate the transcription of
target genes (in a study using a estrogen-dependent breast cancer cell line as
model, 89 such genes were identified).[ Since estrogen enters all cells, its
actions are dependent on the presence of the ER in the cell. The ER is
expressed in specific tissues including the ovary, uterus and breast.
While
estrogens are present in both men and women, they are
usually present at significantly higher levels in women of reproductive age.
They promote the development of female secondary sexual characteristics, such as breasts, and are
also involved in the thickening of the endometrium and other aspects of regulating the
menstrual cycle. In males, estrogen regulates certain functions of the reproductive system important to the maturation of sperm and may be necessary for a healthy libido. Furthermore, there are several other
structural changes induced by estrogen in addition to other functions.
Structural
·
Promote formation of female secondary sex characteristics
·
Accelerate metabolism
·
Increase fat stores
·
Stimulate endometrial growth
·
Increase uterine growth
·
Increase vaginal lubrication
·
Thicken the vaginal wall
·
Maintenance of vessel and skin
·
Reduce bone resorption, increase bone formation
Protein synthesis
·
Increase hepatic production of binding proteins
Coagulation
·
Increase circulating level of factors 2, 7, 9, 10, plasminogen
·
Decrease antithrombin III
·
Increase platelet adhesiveness
Lipid
·
Increase HDL, triglyceride
·
Decrease LDL, fat deposition
Fluid balance
·
Salt (sodium) and water retention
·
Increase cortisol, SHBG
Gastrointestinal tract
·
Reduce bowel motility
·
Increase cholesterol in bile
Melanin
·
Increase pheomelanin, reduce eumelanin
Cancer
·
Support hormone-sensitive breast cancers (see section below)
Lung function
·
Promotes lung function by
supporting alveoli (in rodents
but probably in humans).
Uterus lining
·
Estrogen
together with progesterone promotes and maintains the uterus lining in preparation for implantation
of fertilized egg and maintenance of uterus function during gestation period,
also upregulates oxytocin receptor in myometrium
Ovulation
·
Surge in estrogen level induces
the release of luteinizing hormone, which then triggers ovulation by releasing the egg
from the Graafian follicle in the ovary.
Progestins

Progestins originate from both
ovaries and placenta, and mediate menstrual cycle and maintain pregnancy.
Progesterone has key effects via non-genomic signalling on human sperm as
they migrate through the female tract before fertilization occurs, though the
receptor(s) as yet remain unidentified. Detailed
characterisation of the events occurring in sperm in response to progesterone
has elucidated certain events including intracellular calcium transients and
maintained changes, slow calcium
oscillations, now thought to
possibly regulate motility. Interestingly
progesterone has also been shown to demonstrate effects on octopus spermatozoa.
Progesterone modulates the activity of CatSper (cation channels of sperm) voltage-gated Ca2+ channels. Since eggs release
progesterone, sperm may use progesterone as a homing signal to swim toward eggs
(chemotaxis). Hence
substances that block the progesterone binding site on CatSper channels could
potentially be used in male contraception.
Progesterone is sometimes called the "hormone of
pregnancy", and it has many
roles relating to the development of the fetus:
·
Progesterone
converts the endometrium to its secretory stage to
prepare the uterus for implantation. At the same time progesterone affects the
vaginal epithelium and cervical mucus, making it thick and
impenetrable to sperm. If pregnancy does not occur,
progesterone levels will decrease, leading, in the human, to menstruation. Normal menstrual bleeding is
progesterone-withdrawal bleeding. If ovulation does not occur and the corpus
luteum does not develop, levels of progesterone may be low, leading to anovulatory dysfunctional uterine
bleeding.
·
During
implantation and gestation, progesterone appears to
decrease the maternal immune response to allow for the acceptance of the pregnancy.
·
Progesterone
decreases contractility of the uterine smooth muscle.
·
In addition
progesterone inhibits lactation during pregnancy. The fall in progesterone levels following delivery is
one of the triggers for milk production.
·
A drop in
progesterone levels is possibly one step that facilitates the onset of labor.
The fetus metabolizes placental progesterone in the
production of adrenal steroids.

Androgens

Androgens
originate in the adrenal cortex and gonads and primarily affect maturation and
function of secondary sex organs (male sexual determination).
http://www.youtube.com/watch?v=nLmg4wSHdxQ&feature=fvwrel
The main androgen is testosterone. Its synthesis is regulated by the
luteinizing hormone. Testosterone forms the secondary sexual features in males.
A subset of androgens,
adrenal androgens, includes any of the 19-carbon steroids synthesized by the adrenal cortex, the
inner-most layer of the adrenal cortex (zonula reticularis—innermost region
of the adrenal cortex), that function as weak steroids or steroid precursors,
including dehydroepiandrosterone (DHEA), dehydroepiandrosterone
sulfate (DHEA-S), and androstenedione.
Besides testosterone, other androgens include:
·
Dehydroepiandrosterone (DHEA) is a steroid hormone produced in the adrenal cortex from cholesterol. It is the primary precursor
of natural estrogens. DHEA is also called dehydroisoandrosterone ordehydroandrosterone.
·
Androstenedione (Andro) is an androgenic steroid produced by the testes, adrenal cortex, and ovaries. While androstenediones are
converted metabolically to testosterone and other androgens, they are also the parent structure of estrone. Androstenediol is the steroid metabolite thought to act as the main regulator of gonadotropin secretion.
·
Androsterone is a chemical byproduct created during the breakdown of androgens, or
derived fromprogesterone, that also exerts minor
masculinising effects, but with one-seventh the intensity of testosterone. It
is found in approximately equal amounts in the plasma and urine of both males and females.
·
Dihydrotestosterone (DHT) is a metabolite of testosterone, and a more potent androgen than testosterone in that it
binds more strongly to androgen receptors. It is produced in the adrenal cortex.
Testosterone is the primary androgenic hormone. It
instills its effects on the body both directly, and through its conversion to
metabolites (DHT, estradiol etc). Androgens and other steroid
hormones primarily exert their direct activities through binding to specific
receptors present in the cytosol of cells. Upon binding to the receptor, the
hormone forms a complex that then travels to the nucleus of cells where it
interacts with DNA to promote the formation of specific proteins that then
direct the actual biological changes.
Within the
central nervous system (CNS), androgen receptors are heavily located in
specific places. Androgens and other steroid hormones are able to penetrate the
blood brain barrier and interact with their appropriate CNS cytosolic
receptors. The hypothalamus and anterior pituitary gland are particularly dense
in androgen receptors, and here they help regulate the secretion of androgens
as well as other hormones that control a wide variety of biological functions.
Androgen receptors are also located in parts of the cerebral cortex, medulla,
and amygdala. Here their specific functions are not as well characterized.
The processes
of androgen action that involve receptor binding and DNA translation are known
as receptor mediated, or “genomic”, hormone actions. However, there are also
lesser known actions of steroid hormones that are non-genomic in mechanism.
Non-genomic activities are particularly key in the central nervous system where
they combine with genomic activities to produce specific effects.
Non-genomic
actions of steroid hormones differ in a very important way from genomic
actions. Genomic effects are manifested over a relatively long period of time
(days) because they require a complex cascade of events (binding, translation, transcription,
accumulation of active enzyme products) before the actual physiology of the
target organ is altered. On the other hand, genomic actions are extremely rapid
(<1 minute). They are rapid because their effects involve an immediate
modulation of the membranes of cells (particularly neural cells). These
modulations may include changes to the permeability of the membrane, as well as
effects on the opening of vital ligand gated ion channels. The end result is a
quick and significant influence upon the activities of key areas of the brain,
and the relevance of this to the medicinal use of androgenic hormones or
prohormones should not be overlooked.

Effect of sex
hormones on protein metabolism:
1.
stimulate
the processes of protein, DNA, RNA synthesis;
2.
cause
the positive nitrogenous equilibrium.
Effect
of sex hormones on carbohydrate metabolism:
1. activate the
Krebs cycle;
2. activate the
synthesis of glycogen in liver.
Effect of sex
hormones on lipid metabolism:
1. enhance the
oxidation of lipids;
2. inhibit the
synthesis of cholesterol.
Effect of sex
hormones on energy metabolism:
-
stimulate
the Krebs cycle, tissue respiration and ATP production.
Sex hormones
are used for treatment of variety diseases. For example, testosterone and its
analogs are used as anabolic remedies; male sex hormones are used for the
treatment of malignant tumor of female sex organs and vice versa.
Tissue hormones.
Prostaglandins.
The precursor of prostaglandins is arachidonic acid. Time half-life - 30 s.
There are different prostaglandins and they have a lot of physiological and
pharmacological effects and different prostaglandins have different effects.


Prostaglandins
were first discovered and isolated from human semen in the 1930s by Ulf von
Euler of Sweden. Thinking they had come from the prostate gland, he named them
prostaglandins. It has since been determined that they exist and are
synthesized in virtually every cell of the body.

Prostaglandins,
are like hormones in that they act as chemical messengers, but do not move to
other sites, but work right within the cells where they are synthesized.
Prostaglandins
are unsaturated carboxylic acids, consisting of of a 20 carbon skeleton that
also contains a five member ring. They are biochemically synthesized from the
fatty acid, arachidonic acid.
The unique
shape of the arachidonic acid caused by a series of cis double bonds helps to
put it into position to make the five member ring. See the prostaglandin in the
next panel.
Functions of Prostaglandins:
There are a
variety of physiological effects including:
-
1.
Activation of the inflammatory response, production of pain, and fever. When
tissues are damaged, white blood cells flood to the site to try to minimize
tissue destruction. Prostaglandins are produced as a result.
-
2.
Blood clots form when a blood vessel is damaged. A type of prostaglandin called
thromboxane stimulates constriction and clotting of platelets. Conversely,
PGI2, is produced to have the opposite effect on the walls of blood vessels
where clots should not be forming.
-
3.
Certain prostaglandins are involved with the induction of labor and other
reproductive processes. PGE2 causes uterine contractions and has been used to
induce labor.
-
4.
Prostaglandins are involved in several other organs such as the
gastrointestinal tract (inhibit acid synthesis and increase secretion of
protective mucus), increase blood flow in kidneys, and leukotriens promote
constriction of bronchi associated with asthma.
Effects of Aspirin
and other Pain Killers:
When you see
that prostaglandins induce inflammation, pain, and fever, what comes to mind
but aspirin. Aspirin blocks an enzyme called cyclooxygenase, COX-1 and COX-2,
which is involved with the ring closure and addition of oxygen to arachidonic
acid converting to prostaglandins. The acetyl group on aspirin is hydrolzed and
then bonded to the alcohol group of serine as an ester. This has the effect of
blocking the channel in the enzyme and arachidonic can not enter the active
site of the enzyme.

By inhibiting
or blocking this enzyme, the synthesis of prostaglandins is blocked, which in
turn relives some of the effects of pain and fever.
Aspirin is
also thought to inhibit the prostaglandin synthesis involved with unwanted
blood clotting in coronary heart disease. At the same time an injury while
taking aspirin may cause more extensive bleeding.
See the
following chime tutorial for the detailed molecular basis for the inhibition of
the COX enzyme by aspirin.
Kallicrein-kinin
system. Kinins -
group of peptides with similar structure and biological properties. The main
kinins - bradykinin and kallidine.
Kinins are
formed from their precursors kininogens that
are synthesized in liver owing to acting of kallicreins. Kallicreins are also
formed from inactive precursors prekallicreins by means of proteolysis.
Functions: -
kinins relax the smooth muscles of blood vessels and decrease the blood
pressure;
-
increase
the capillaries permeability;
-
takes
part in the inflammatory processes.
Bradykinin is
a potent endothelium-dependent vasodilator, causes
contraction of non-vascular smooth muscle, increases
vascular permeability and also is involved in the mechanism
of pain. Bradykinin
also causes natriuresis,
contributing to a drop in blood pressure.

Bradykinin
raises internal calcium levels in neocortical astrocytes causing them to release glutamate.
Bradykinin is
also thought to be the cause of the dry cough in some patients on angiotensin converting enzyme (ACE) inhibitor drugs. It is thought
that bradykinin is converted to inactive metabolites by angiotensin converting enzyme (ACE),
therefore inhibition of this enzyme leads to increased levels of bradykinin
which causes a dry cough. This refractory cough is a common cause for stopping ACE inhibitor therapy. In which
case angiotensin II receptor antagonists (ARBs) are the next line of treatment.
Renin-angiotensin system. Renin - enzyme
that is synthesized in special cells located near the renal glomerules.
Renin acts on angiotensinogen. As result angiotensin-I
is formed. Under the effect of peptidase
angiotensin-I is converted to angiotensin-II.
Angiotensin-II causes 2 effects:

-
narrows
the vessels and increases the blood pressure;
-
stimulates
the secretion of aldosterone.
The decrease of renal blood stream is
the specific stimulant for renin secretion.
{kind=link}