 4.1. Хромосомнi захворювання
4.1. Хромосомнi захворювання
|
4.1. Хромосомнi захворювання
|
ХРОМОСОМНI ЗАХВОРЮВАННЯ
До хромосомних захворювань вiдносять форми патологiї, що клiнiчно проявляються множинними вродженими вадами розвитку, генетичним грунтом яких є вiдхилення вiд нормального вмiсту в клiтинах органiзму кiлькостi хромосомного матерiалу, тобто обумовленi геномними чи хромосомними мутацiями.
Бiльшiсть хромосомних захворювань мають спорадичний характер, тобто виникають внаслiдок геномної (хромосомної) мутацiї в гаметi здорового батька або матерi, чи в перших подiлах зиготи, а не успадковуються в поколiннях, що пов'язано з високою смертнiстю хворих в дитячому вiцi, а також статевим недорозвитком i нездатнiстю давати потомство. Фенотипiчну основу хромосомних захворювань формують порушення раннього ембрiонального перiоду. Саме тому патологiчнi змiни складаються ще в пренатальному перiодi розвитку органiзму i або обумовлюють загибель ембрiона чи плода, або створююють основну клiнiчну картину захворювання вже в новонароджених. Виняток становлять аномалiї статевого розвитку, що формуються в основному в перiодi статевого дозрiвання.
Сучасний перiод вiдрiзняється видiленням цитогенетично i клiнiчно бiльш рiзних синдромiв, що обумовленi дисбалансом по окремих сегментах практично всiх 22 аутосом i 2 статевих хромосом. Роль хромосомної патологiї в загибелi ембрiона i плода у людини в загальнiй кiлькостi множинних вад розвитку у новонароджених велика. В середньому близько 40 % дiагностованих спонтанних абортiв обумовленi хромосомними порушеннями, близько 6 % всiх мертвонароджених мають змiни хромосомного апарату. На 1000 мертвонароджених немовлят 3-5 мають хромосомнi хвороби. Якщо ж всi випадки множинних вроджених вад прийняти за 100 %, то 35-40 % будуть припадати на порушення стану хромосом.
Все це визначає необхiднiсть знання акушерами, педiатрами, дитячими ендокринологами, сiмейними лiкарями, психоневрологами, ортопедами, патологоанатомами i iншими спецiалiстами етiологiї, клiнiчної картини, принципiв дiагностики, лiкування i профiлактики цiєї великої групи патологiчних станiв.
Дiагностика спадкових захворювань повинна базуватися на основi генеалогiчного аналiзу (ретельного вивчення сiмейного анамнезу декiлькох поколiнь), близнюкового методу, цитологiчних дослiджень, методiв бiохiмiчної генетики, дерматоглiфiки, ендокринологiчних методiв, а головне - на основi виявлення типових симптомiв спадкових захворювань.
Основним носiєм спадкових властивостей живих органiзмiв є хромосомний аппарат ядра клiтини. Для кожного бiологiчного виду характерний постiйний хромосомний комплекс, який складається iз визначеної кiлькостi хромосом. Так, для одного з видiв круглих червiв (кiнська аскарида) типова наявнiсть 2 хромосом, тодi як для деяких ракоподiбних характерна наявнiсть 200 хромосом. У бiльшостi вищих органiзмiв кожна клiтина мiстить диплоїдний (2 n) хромосомний набiр. Хромосоми вiдрiзняються одна вiд одної формою i розмiрами. Сукупнiсть кiлькiсних i якiсних ознак хромосом, що виявляється мiкроскопiчно в поодинокiй клiтинi, називається карiотипом.
Нормальне диплоїдне число хромосом у людини дорiвнює 46. Через недосконалiсть цитологiчної технiки загальне число хромосом у людини довго (з 1912 до 1956 року) вважали рiвним 48. I лише в 1956 роцi шведськi цитологи Н.Tjio i A. Levan застосовували удосконалену цитологiчну методику, довели, що модельна кiлькiсть хромосом у людини дорiвнює 46. Цi данi були пiдтвердженi в тому ж роцi англiйськими вченими С.Е. Ford i J.L. Hamerton. З цього часу почався бурхливий розвиток цитогенетики.
Хромосомний комплекс людини складається з 23 пар. З них 22 пари аутосом, а 23-я пара - статевi хромосоми, основна роль яких - у визначеннi статi. Так, жiноча стать представлена двома Х-хромосомами - ХХ, а чоловiча - ХY-хромосомами. Слiд вiдмiтити, що багато спадкових захворювань як генних, так i хромосомних, пов'язанi iз змiнами в статевих хромосомах (Х i Y).
46 хромосом мiстять соматичнi клiтини. Статевi клiтини мають хромосомний набiр вдвiчi менший - 23 хромосоми. Аутосоми однаковi як у чоловiкiв, так i у жiнок.
Загальна семiотика хромосомних патологiй
Майже всi хромосомнi захворювання супроводжуються багаточисленними ураженнями скелету, порушеннями психiки, вродженими вадами зовнiшнiх i внутрiшнiх органiв, затримкою росту, ураженням нервової, ендокринної та iнших систем, зниженою регенераторною функцiєю, пiдвищеною захворюванiстю i смертнiстю.
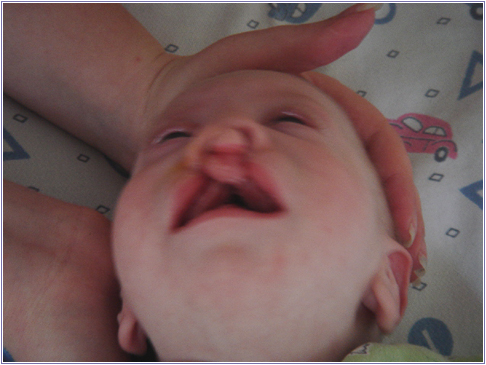
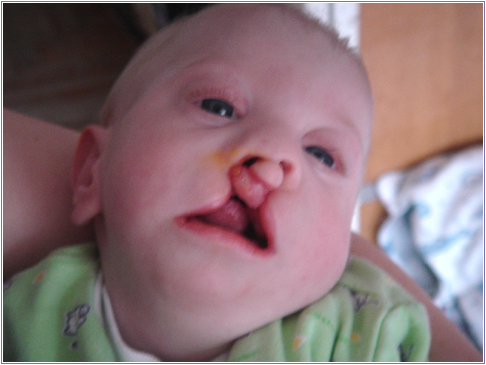
Рис.1. Незарощення м'якого пiднебiння.
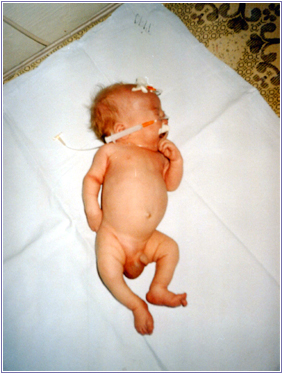
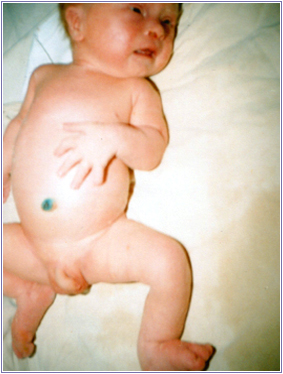
Рис.2. Множиннi дисморфiї у 2-мiсячної дитини.
Дiагностичнi ознаки хромосомних аномалiй можна роздiлити на 3 групи:
А - комплекс ознак, що дозволяють запiдозрити хромосомну аномалiю. Це загальнi ознаки: фiзичний недорозвиток, ряд дизморфiй мозкового i лицьового черепа (рис.1), клишоногiсть(рис.2), ряд аномалiй внутрiшнiх органiв (серця, легень, нирок).
В - ознаки, якi зустрiчаються в основному при певних хромосомних захворюваннях. Їх комбiнацiя дозволяє в бiльшостi випадкiв дiагностувати хромосомну аномалiю. Зокрема, при трисомiї 18-ї хромосоми (хвороба Едвардса) - це долiхоцефалiя (89,6 %), флексорне положення кистей (96,1 %), "стопа-качалка" (76,2 %), короткий i широкий палець стопи (70,6 %). При трисомiї по 13-й хромосомi (хвороба Патау) - це щiлина верхньої губи i пiднебiння (68,7 %), флексорне положення кистей (44,4 %), косоокiсть (31,4 %), дефект скальпа (30,5 %).
С - ознаки, характернi лише для окремих хромосомних аномалiй: крик кiшки при синдромi 5 р; алопецiя при синдромi 18 р.
Вважають, що 0,5 % всiх новонароджених мають хромосомнi аномалiї. Приблизно 1:400 хлопчикiв i 1:600 дiвчаток мають порушення статевих хромосом..Серед них найчастiше зустрiчаються синдром Клайнфельтера i Шерешевського-Тернера. Серед аномалiй аутосом найчастiше зустрiчається хвороба Дауна 1: 700-1:800, iншi --в декiлька разiв рiдше. У недоношених дiтей хромосомнi аномалiї зустрiчаються в 4 рази частiше, нiж у доношених. Ще бiльше хромосомних захворювань в матерiалi абортiв - в середньому 40 %. До аномалiй статевих хромосом вiдносяться синдроми Клайнфельтера, Шерешевського-Тернера, трисомiї - Х i iнших.
Синдром Клайнфельтера - спостерiгається у осiб чоловiчої статi, що обумовлено порушенням статевих хромосом в бiк збiльшення кiлькостi Х-хромосом. Синдром вiдносять до первинних форм чоловiчого гiпогонадизму.
Поширенiсть: 1.13 випадкiв на 1000 хлопчикiв.
Етiопатогенез: В основi захворювання лежить патологiчний комплекс статевих хромосом. Замiсть нормального карiотипу з 46-а хромосомами при синдромi Клайнфельтера є 47 хромосом. У осiб чоловiчої статi при цьому є двi Х-хромосоми (замiсть однiєї Х i однiєї Y в нормi), тобто набiр статевих хромосом - ХХY. Крiм цього, при даному синдромi можлива рiзноманiтнiсть цитогенетичних варiантiв i їх поєднання. Виявлено декiлька типiв полiсомiї по Х i Y- у осiб чоловiчої статi: 47 XXY; 49 XXXY; 49 XXXXY; 47 XYY; 48 XYYY; 48XXYY; 49 XXXYY. Найбiльш поширений полiсомний по Ч-хромосомi синдром Клайнфельтера (ХХY).
Клiнiка синдрому найбiльш виразна у перiод статевого дозрiвання. У хлопчикiв вiдмiчається високий зрiст, подовженi кiнцiвки, євнухоїдна будова тiла, слабо розвинена мускулатура, а також атрофiя яєчок, недостатнiй розвиток вторинних статевих ознак. Часто в перiод стетевого дозрiвання з'являється гiнекомастiя (збiльшення грудних залоз). Як правило, вiдмiчаються рiзнi ступенi розумової вiдсталостi i психiчних захворювань - шизофреноподiбнi стани, манiакально-депресивний психоз i iншi. У осiб 3-4 Х-хромосомами спостерiгаються скелетнi аномалiї, бiльш вираженi гiпогенiталiзм, крипторхiзм, розумова неповноцiннiсть, аж до iдiотiї. Обмiнних порушень при цьому захворюваннi не встановлено. Серцево-судинна дiяльнiсть не порушена. Вiдмiчається вмiст хорiогонадотропiну. Дiагноз базується на клiнiчних даних, а також на визначеннi патологiчного карiотипу, може бути пiдтверджений при дослiдженнi статевого хроматину в клiтинах. В зв'язку з наявнiстю додаткової Х-хромосоми визначається статевий хроматин, якого в нормi в клiтинах у осiб чоловiчої статi немає.
Дiагностичнi критерiї: гiпогенiталiзм, гiпогонадизм, характернi аномалiї карiотипу.
Своєрiдним рiзновидом синдрому Клайнфельтера є полiсомiя по Y-хромосомi (XXXY). Вперше його описав Ж.С. Хаушка в 1962 роцi у фенотипiчно нормальних чоловiкiв. Частота даного синдрому в нормальнiй популяцiї втасновлена в межах 0,1-0,15 % , проте в популяцiї психiчно хворих вона значно вища (вiд 0,45 до 15 %).
Клiнiчно синдром XYY нагадує синдром Клайнфельтера. Однак у чоловiкiв з хромосомним комплексом XYY зрiст значно вищий - в середньому вiн бiльше 180-185 см. Iнтелект збережений, але розумовий розвиток вiдповiдає низьким чи середнiм показникам. Частина з них вiдрiзняється агресивною поведiнкою i олiгофренiєю. Будь-яких специфiчних соматичних вiдхилень у бiльшостi з цих людей немає, тому вони рiдко потрапляють в поле зору лiкаря. Як i при синдромi Клайнфельтера, у хворих з XYY симптомокомплексом спостерiгається безплiддя, ендокринний дисбаланс i гiпоплазiя яєчок. Гiстологiчно виявляється зменшення термiнальних клiтин сiм'яних канальцiв, гiалiнiзацiя i потовщення базальних мембран.
Лiкування синдрому Клайнфельтера. В силу того, що хвороба проявляється в перiод пубертату, основним iз проявiв якої є статевий iнфантилiзм, пацiєнтам призначають препарати чоловiчих статевих гормонiв: пропiонат тестостерона внутрiшньом'язово 2-3 рази в тиждень. В легких випадках статевого недорозвитку використовують метилтестостерон по 10 мг 2-3 рази на добу сублiнгвально. Однак, слiд пам'ятати, що пацiєнтам iз глибоким розумовим недорозвитком, терапiя андрогенами не проводиться, оскiльки вона може спонукати до асоцiальної поведiнки. Помiрна гiнекомастiя купується застосуванням статевих чоловiчих гормонiв, а в разi випадкiв показане хiрургiчне лiкування.
Медико-генетичне консультування. Високим ризиком є вiк майбутньої мами старше 35 рокiв. В сiм'ї де є дитина iз синдромом Клайнфельтера випадки повторного народження хворого хлопчика рiдкiснi.
Прогноз для життя сприятливий; хлопчики безплiднi.
Пренатальна дiагностика. Методи масової скринiнгової дiагностики не iнформативнi. При селективнiй дiагностицi (за показами) враховують данi УЗД плода, амнiоцентезу, визначення карiотипу плода.
Синдром Шерешевського - Тернера - клiнiчний симптомокомплекс, який спостерiгається у дiвчат. Характеризується своєрiдним фiзичним розвитком, вiдставанням в статевому розвитку.
Поширенiсть: 1 випадок на 7700 або 1 випадок на 3000-3500 новонароджених дiвчаток.
Етiопатогенез: Захворювання описане в 1925 р. ендокринологом Н.А. Шерешевським, який вважав, що воно пов'язане з недорозвитком передньої частки гiпофiза i яйникiв в комбiнацiї з iншими вродженими аномалiями розвитку. Класичний опис належить Х.Х. Тернеру (1938). В 1954 р. Polani i спiвавтори встановили вiдсутнiсть типового для жiночої статi статевого хроматину у цих хворих i передбачили, що комплекс статевих хромосом вiдповiдає чоловiчому типу - ХY. Цитогенетично синдром ХЩ вiдкрив С.Е. Форд в 1959 роцi.
Синдром Тернера у чоловiкiв описав Флавей. Хворi з чоловiчим синдромом Шерешевського-Тернера фенотипiчно хлопчики карликового зросту з короткою шиєю, аплазiєю яєчок, дисгенезiєю канальцiв i зниженим iнтелектом. Y-хромосома зберiгає до деякого ступеня свою активнiсть. Крiм того, у дiвчат з недорозвитком гонад, у хлопчикiв з гермафродитизмомо i частими пухлинами статевих залоз визначається мозаїчний хромосомний набiр ХО/ХY, всi вони хроматин-негативнi, як i iншi варiанти синдрому Шершевського-Тернера з хромосомними наборами ХО, 46 ХY, ХО/ХY i т.д.
При рiзних видах мозаїцизму клiнiчнi ознаки у цих хворих менш вираженi, нiж при синдромi ХО (набряк при народженнi i крилоподiбна шийна складка вiдмiчається в окремих випадках, зрiст хворих може залишатись нормальним, бiльш частi спонтаннi менструацiї). Хроматин-позитивнi варiанти синдрому Шерешевського-Тернера представленi карiотипами ХО/ХХ/ХХХ, ХО/ХХХ, дохромосомним мозаїцизмом ХХ1 i ХХ/ХХ1, карiотипом з кiльцевою Х-хромосомою: ХО/ХХ2, ХО/ХХ/ХХ2 , ХО/ХХ2/ ХХ2/Х2 i статевою хромосомою з делецiєю довгого плеча Х1ХО/ХХ. Гонади таких хворих недорозвинутi, виражена iнфантильнiсть.
В основi захворювання лежить патологiчний набiр хромосом (45 хромосом). Статевi хромосоми представленi однiєю Х-хромосомою (комплекс позначається ХО). В рядi випадкiв можуть бути морфологiчнi змiни в Х-хромосомi при нормальному кiлькiсному вiдношеннi карiотипiв. Крiм того, є хворi з карiотипом, характерним для синдрому Шерешевського-Тернера - ХО-хромосоми визначаються лише в частинi клiтин, а в iнших є iншi карiотипи - ХХ, XY, ХХХ, ХХY, тощо. Близько 80% хворих хроматин-негативнi. Таких хворих називають "мозаїками". Вiдсутнiсть або змiна Х-хромосоми призводить до порушення синтезу бiлкiв i ферментiв, що визначає порушення обмiну в органiзмi i призводить до багаточисленних аномалiй.
Клiнiка. Найбiльш частим симптомом є низький зрiст. Ще в дитинствi цi хворi вiдстають вiд ровесникiв i до моменту статевого дозрiвання їх зрiст складає 130-145 см. Iснують данi про велику частоту синдрому Шерешевського-Тернера серед низькорослих дiвчаток в Японiї. Другою характерною ознакою є статевий iнфантилiзм. Це особливо часто проявляється в пубертатному перiодi у виглядi аменореї, недорозвитку статевих органiв i вторинних статевих ознак. На мiсцi яйникiв визначаються тяжi. Гiстологiчне дослiдження їх виявляє наявнiсть сполучної тканини, в якiй зустрiчаються поодинокi острiвцi тканини яйника з премордiальними i дуже рiдко з розвинутими фолiкулами. Цi хворi безплiднi. Будова тiла непропорцiйна - довжина верхньої половини тулуба значно бiльша нижньої. Вуха деформованi, низько розмiщенi. Тверде пiднебiння iнколи високе i вузьке ("готичне"), вiдмiчається неправильний рiст зубiв. Шия широка i коротка. Вiдмiчається низький рiст волосся на шиї. Широкi шкiрнi складки на шиї, що йдуть вiд соскоподiбних вiдросткiв до плечей, надають їй типового вигляду крилоподiбної шиї (pterigium coli). Аномалiї кистi проявляються у вкороченнi четвертих (за рахунок коротких метакарпальних кiсток) i викривлення п'ятих пальцiв. III, IУ, У пальцi стоп також вкороченi i деформованi. Часто вiдстань мiж I i II пальцями стопи збiльшена. Вiдмiчається стiйкий набряк кiнцiвок. При синдромi Шерешевського-Тернера є ряд змiн з боку внутрiшнiх органiв - вродженi вади серця i великих судин (коарктацiя аорти, незарощення мiжшлуночкової перетинки (рис.3), стеноз устя легеневої артерiї (рис.4)), аномалiї нирок (пiдковоподiбна нирка, подвiйнi мисочки або сечоводи).
В неврологiчному статусi патологiчних змiн немає. В 50 % випадкiв хворi з синдромом Шерешевського-Тернера розумово вiдсталi. Вони пасивнi i астенiчнi, схильнi до психогенних реакцiй i реактивних психозiв. Часто спостерiгається порушення слуху (близько 40 %), спостерiгаються отити. Це пояснюється аномальним розмiщенням слухової трубки через неправильне формування каудального вiддiлу зовнiшнього слухового проходу.
Дiагноз синдрому Шерешевського-Тернера у новонароджених поставити важко, оскiльки ще не проявляються характернi ознаки. Проте найбiльш характернi ознаки у виглядi короткої шиї з надлишком шкiри i шкiрними складками, лiмфатичного набряку стоп, гомiлок, кистей рук i передплiч присутнi, що дозволяє запiдозрити дану хромосомну патологiю. В шкiльному вiцi i особливо у пiдлiткiв проявляється вiдставання в ростi, слабкий розвиток статевих ознак, аменорея, характерний зовнiшнiй вигляд. З вiком дiагноз грунтується на клiнiчнiй картинi i визначеннi статевого хроматину. Визначення патологiчного карiотипу (45 хромосом при статевому наборi ХО тощо), вiдсутнiсть статевого хроматину або недостатня кiлькiсть його пiдтверджують дiагноз захворювання.

Рис.3. Незарощення мiжшлуночкової перетинки.
Рис.4. Стеноз устя легеневої артерiї.
Дiагностичнi критерiї: статевий iнфантилiзм, первинна аменорея, низький рiст, шкiрнi складки на шиї, вродженi вади серцево-судинної, сечовидiльної систем) розвитку.
Лiкування синдрому Шерешевського-Тернера проводиться етапно. На першому етапi використовують анаболiчнi препарати, спрямованi на збiльшення росту. На другому - з 14-15 рокiв проводиться замiсна терапiя естрогенами - спочатку постiйно до появи першої mensis, а далi курсами, що iмiтують нормальний менструальний цикл.
Медико-генетичне консультування. Повторне народження у сiм'ї дитини iз синдромом Шерешевського-Тернера дуже рiдкiсне.
Прогноз для життя в цiлому сприятливий, за винятком тяжких вад серця, ренальної гiпертензiї.
Пренатальна дiагностика. Масовий скринiнг дає можливiсть при 14-18 тижневiй вагiтностi виявити наступнi змiни при УЗД плода: шийно-потиличну цистогiгрому; крилоподiбнi складки на шиї та її вкорочення, лiмфатичний набряк м'яких тканин, гiпертелоризм. В цьому ж термiнi у кровi вагiтної та амнiотичнiй рiдинi пiдвищується вмiст альфа-протеїну. Вище наведенi факти потребують обов'язкового медико-генетичного консультування для вирiшення питання про народження дитини.
Аномалiї аутосом
Аномалiї аутосом можуть бути рiзного типу - трисомiї, моносомiї, а також порушення структури окремих хромосом (транслокацiї, делецiї, фрагментацiї, кiльцеподiбнi хромосоми тощо), мозаїцизм. Порушення у великих i середнiх хромосомах в рехультатi нерозходження в мейозi i утворення трисомiї призводять до ураження плода, несумiсного з життям, i є причиною спонтанних абортiв i мертвонароджень. Це має мiсце в 6-8 % спонтанних абортiв. Особливо частi хромосомнi аберацiї у спонтанних викиднiв перших трьох мiсяцiв вагiтностi. Найбiльш часто при кiлькiсному порушеннi хромосом зустрiчаються трисомiї. В постнатальному перiодi зi збереженням життя хоча б на короткий перiод часу сумiснi повнi трисомiї по небагатьох хромосомах: 8, 9, 13, 18, 21 i 22, причому по аутосомах 8 i 9 частi випадки мозаїцизму, що без сумнiвну зменшує летальний ефект дисбалансу генетичного матерiалу.
Повна моносомiя у новонароджених - виключно рiдке явище, не доведене кiнцево навiть для найбiльш цiнної хромосоми 21. Повнi моносомiї очевидно нежиттєздатнi вже на стадiї гамет i зиготи по всiх аутосомах, оскiльки навiть в матерiалi спонтанних абортiв описання таких знахiдок поодинокi i вiдомi вiдносно лише до деяких аутосом.
Структурнi перебудови притаманнi всiм без винятку аутосомам. По бiльшостi аутосом вдалося виявити вiдносну клiнiчну специфiчнiсть фенотипiчних вiдхилень порушень будови хромосоми, проте спостереження i накопичення досвiду ще не закiнчено, оскiльки природа невичерпна в утвореннi все нових i нових випадкiв патологiї, вирiшальна здатнiсть цитогенетичних методiв безперервно пiдвищується.
Синдром Патау
Синдром Патау - симптомокомплекс множинних аномалiй, обумовлених трисомiєю 13 хромосоми.
Повна трисомiя хромосоми 13 була описана в 1960 роцi майже зразу пiсля вiдкриття трисомiї 21 i з того часу залишається другою за частотою повною аутосомною трисомiєю. Дiти частiше народжуються у матерiв старшого вiку.
Поширенiсть: 1 випадок на 7800-8000 народжених, з незначною перевагою народжуються хворi дiвчатка.
Етiологiя. Описанi 2 цитогенетичних варiанти синдрому Патау: проста трисомiя (13 d) або робертсонiвська транслокацiя, якi зустрiчаються у спiввiдношеннi 5,6:1. Фенотипiчно обидвi форми iдентичнi.
Клiнiка. Зовнiшнiй вигляд хворого з синдромом досить специфiчний. Новонародженi мають нормальну масу тiла i нормальнi розмiри. Клiнiчно у них спостерiгаються симптоми недорозвитку центральної нервової системи, мiкроцефалiя, неправильно сформованi низько розмiщенi вуха, аномалiї очного яблука (мiкрофтальмiя i анофтальмiя), одно- чи двобiчне незарощення губи i пiднебiння, полiдактилiя, пiдвищена рухливiсть суглобiв, вродженi вади внутрiшнiх органiв (серцево-судинної i сечовидiльної систем, травного каналу), часто спостерiгаються судоми. З iнших клiнiчних симтомiв слiд вiдмiтити гемангiоми пальцiв кистi, деформацiю стопи, пупковi i пахово-калитковi грижi, крипторхiзм, глухоту. Глухота у хворих з трисомiєю 13 зустрiчається у 80 % випадкiв. Найчастiше змiненi середнє вухо i нижня частина внутрiшнього вуха. Дiагностичнi критерiї: комплекс множинних аномалiй: черепно-лицевi дисморфiї: мiкро- та тригоноцефалiя, гiпотелоризм, розщелина губи та пiднебiння, дефекти скальпа, вади кiнцiвок ( флексорне положення кистей, полiдактилiя, стопи - качалки); аномалiї ц.н.с. та серцево-судинної системи, статевих органiв. Обов'язкове цитогенетичне пiдтвердження дiагнозу.
Лiкування. Множиннi аномалiї та вади зводять на нiвець будь-якi лiкувальнi заходи.
Медико-генетичне консультування. У випадку простої трисомiї ризик для наступних дiтей низький (менше 1 %).
Прогноз несприятливий: 2/3 дiтей помирають у перинатальному перiодi; близько 1/3 помирає на 1 роцi життя.
Пренатальна дiагностика. Пiд час масового УЗД при термiнi 16-20 тижнiв вагiтностi виявляють множиннi вади розвитку. За таких обставин є реальна потреба пренатальної дiагностики iз проведенням амнiоцентезу i карiотипування клiтин плода.
Деяким вагiтним рекомендована селективна дiагностика за умови: вiк майбутньої мами старше 30 рокiв, багатовiддя; гiпотрофiя плода; один iз батькiв - носiй збалансованої транслокацiї 13 d. Саме їм у триместрi вагiтностi проводять амнiоцентез та визначення карiотипу плода.
Синдром Едвардса
Синдром Едвардса - симптомокомплекс множинних аномалiй (зовнiшннiх та вiсцеральних) обумовлених хромосомною патологiєю 18 пари. За частотою серед новонароджених трисомiя 18 стоїть на третьому мiсцi пiсля трисомiй 21 i 13 пар, обумовлюючи рано виявлений i добре вивчений синдром Едвардса.
Поширенiсть: 1 випадок на 800 народжених.
Етiологiя. Трисомiя 18 є майже у всiх випадках результатом простого нерозходження хромосоми, як правило, на стадiї гамети, але часом i на стадiї зиготи (мозаїцизм). Транслокацiйнi форми виключно нечастi.
Клiнiка: Для синдрому характерний рiзкий пренатальний недорозвиток i багаточисленнi вади кiсткової системи, зокрема, кiсток обличчя, але не рiзко вираженi. Характернi є вiдхилення в дерматоглiфiчному рисунку, iз вад внутрiшнiх органiв часто спостерiгаються: шлунково-кишкового тракту (дивертикул Меккеля, пiлоростеноз, атрезiя жовчних шляхiв, ануса), недорозвитк легень, дефект мiжшлуночкової перетинки, вади розвитку клапанiв аорти i легеневої артерiї, вади розвитку статевих органiв, патологiя органiв зору.
Дiагностичнi критерiї: пренатальна гiпоплазiя, множиннi аномалiї розвитку та стигми дисембрiогенезу, серед яких : антимонголоїдний розрiз очей, низько посадженi деформованi вушнi раковини, мiкрогенiя, єдина пупкова артерiя.
Лiкування : здебiльшого симптоматичне, неефективне.
Медико-генетичне консультування. Визначення ризику повторного народження хворої дитини проводять iз врахуванням генеалогiчного, акушерського анамнезiв та карiотипу батькiв.
Прогноз несприятливий: 60 % дiтей помирає у вiцi до 3 мiсяцiв; до рiчного вiку доживає менше 10 % хворих.
Пренатальна дiагностика. Масове УЗД вагiтної у I триместрi може виявити: багатовiддя, гiпотрофiю плода, множиннi аномалiї. Селективна дiагностика показана у випадку: один iз батькiв є носiєм.
Хвороба Дауна
Хвороба Дауна - вроджене захворювання, що обумовлене хромосомною аберрацiєю, в основному 21-ї пари хромосом, що проявляється рядом фiзичних недолiкiв, нерiдко недоумкуватiстю i рiзким зниженням резистентностi до iнфекцiйних захворювань. Хвороба описана в 1866 роцi Down. Серед розумово вiдсталих дiтей цi хворi складають 10-12 %.
Поширенiсть: 1 випадок на 1550 народжених.
Етiологiя i патогенез хвороби протягом багатьох рокiв були не з'ясованi. Вважалось, що в основi хвороби лежить полiгландулярна недостатнiсть, тобто порушення функцiї кiлькох залоз внутрiшньої секрецiї (щитоподiбної залози, гiпофiза i iнших ), рiзке вiдставання в розвитку ЦНС. В 1959 роцi була встановлена iстинна суть захворювання. Виявилось, що при хворобi Дауна найчастiше (в 94% випадкiв ) є трисомiя 21-ї пари хромосом. Таке порушення карiотипу (трисомiя) вiдбувається в тому випадку, коли у матерi при дозрiваннi статевої клiтини пiд впливом часто невiдомих причин вiдбулося нерозходження 21-ї пари хромосом i утворилась яйцеклiтина, що мiстить 24 хромосоми. При заплiдненнi такої яйцеклiтини нормальним сперматозоїдом, що мiстить 23 хромосоми, розвивається дитина з 47 хромосомами, у якої виявляється хвороба Дауна. Дiти з хворобою Дауна, що мiстить в карiотипi 47 хромосом, народжуються переважно у матерiв похилого вiку (в 20 рокiв - 1 випадок на 700, понад 45 - 1 на 20-45 новонароджених). Менша частина хворих (3-5 %) має карiотип з 46 хромосом, при чому одна з них (переважно в 13, 15-й або 22-й парах) має зайву 21 -у хромосому (46 хромосом з транслокацiєю), а також мозаїчнi трисомiї, хромосомнi транслокацiї - 13-15/21, 21/22, 2/21, 4-5/21, 20/21. Хворi з таким карiотипом народжуються переважно у молодих матерiв, якi є носiями такої ж транслокацiї, але мають всього 45 хромосом. Звичайно хромосомна аберацiя виникає не у матерi хворого, а у будь-кого iз попереднiх поколiнь. Клiнiчних вiдмiнностей в перебiгу хвороби Дауна при трисомiї або транслокацiї хромосом не встановлено. При стертих формах захворювання встановлена мозаїка карiотипу, при цьому частина клiтин мiстить нормальний карiотип, а частина клiтин -зайву 21-у хромосому.
Питання про причини виникнення хромосомних аберацiй, що обумовлюють хворобу Дауна, остаточно не з'ясованою. У матерi як до, так i пiсля народження дитини з хворобою Дауна спостерiгається пiдвищена частота спонтанних викиднiв i мертвонароджень. Якщо народжуються рiзнояйцевi близнята, то наявнiсть синдрому Дауна визначається переважно у одного з них; при народженнi однояйцевих близнят хвороба Дауна спостерiгається у двох. Хромосомнi аберацiї можуть бути також обумовленi рядом вiдомих мутагенних факторiв - iонiзуючою радiацiєю, хiмiчними, термiчними впливами тощо.
Клiнiка. Ознаки, що дозволяютиь дiагностувати захворювання в типових випадках виявляються на раннiх етапах життя дитини. Звертає на себе увагу малий зрiст дитини, маленька кругла голова iз скошеною потилицею, iз своєрiдним обличчям - бiдна мiмiка, косий розрiз очей iз складкою бiля внутрiшнього кута (епiкантус), нiс з широким плоским перенiссям, маленькi деформованi вушнi раковини. Рот переважно напiввiдкритий, язик товстий неповороткий, пiднебiння високе, нижня щелепа iнколи виступає до переду. На щоках часто вiдмiчається суха екзема, внаслiдок чого вони гiперемованi. Спостерiгається вкорочення кiнцiвок, особливо в дистальних вiддiлах. Кисть плоска, пальцi рук широкi, короткi. Мiзинець вкорочений, нерiдко серпоподiбно викривлений до середини. Часто вiдмiчається великий промiжок мiж I i II пальцями. Спостерiгається своєрiдний рисунок шкiрних складок на долонях. В нормi серед багаточисленних складок на долонi видiляються 3 найбiльшi - одна з них у виглядi дуги оточує пiдвищення великого пальця, двi iншi перетинають долоню в поперечному напрямку. При хворобi Дауна обидвi поперечнi складки зливаються i утворюють одну поперечну, що йде через всю долоню. Вона буває на обох руках, але частiше добре виражена на однiй. Тiм'ячка при народженнi дуже великi, пiзно закриваються. Iснує три тiм'ячка на тiменi. Рiзко виражена м'язова гiпотонiя.
У фiзичному розвитку дiти вiдстають, але нерiзко. Нервово-психiчний розвиток значно сповiльнений. Вони пiзно починають тримати голову (переважно не ранiше 6 мiсяцiв), сiдати i стояти. Особливо пiзно i погано розвивається мова. Часто у вiцi вiд 2 до 6 рокiв вони вимовляють лише окремi слова, запас слiв невеликий. Пiзнiше дiти вимовляють окремi короткi фрази, але зв'язною мовою вони переважно нiколи не володiють.
З вiком виявляється ряд нових рис захворювання. Голос стає грубим, спостерiгається короткозорiсть, косоокiсть, кон'юнктивiт, блефарит, вогнища депiгментацiї. На периферiї райдужки бiлi плями. Гiпертрофiя сосочкiв язика, неправильний рiст зубiв, карiєс. Виявляються аномалiї будови грудини, iнколи асиметричне розмiщення соскiв. Пупок завжди виступає над поверхнею шкiри. Епiкантус з вiком зникає, м'язова гiпотонiя може наростати.
Розумова вiдсталiсть при хворобi Дауна характеризується переважно iмбецильнiстю рiзних ступенiв, однак iнколи спостерiгається дебiльнiсть, iдiотiя. Деяких дiтей вдається навчити читати i писати, але рахувати, як правило, вони не можуть. Дiти з хворобою Дауна ласкавi, добродушнi, слухнянi, але часом впертi. Вони мають властивiсть до iмiтацiй (наслiдування), але зовсiм не можуть пристосуватись до будь-якої систематичної працi. Спостерiгається вiдставання у статевому розвитку.
У дiтей з хворобою Дауна в 30-40 % випадкiв виявляються вродженi вади серця та судин (незарощення овального отвору, дефект мiжшлуночкової перетинки тощо). В рядi випадкiв спостерiгаються вади розвитку кишечника або iншi вродженi аномалiї. Iнфекцiйнi захворювання у цих дiтей перебiгають дуже важко i ранiше призводили до летальних наслiдкiв.
У дiтей з хворобою Дауна у 15 разiв частiше, нiж в iнших дiтей зустрiчається гострий лейкоз. Iснують данi про поєднання вродженого лейкозу i хороби Дауна, описанi сiмейнi випадки лейкозу. Вважається, що у виникненнi гострих лейкозiв вiдiграють роль геннi мутацiї. Зустрiчається рецесивний тип успадкування з високою пенетрантнiстю гена i домiнантний - з низькою. Родичi хворих з лейкозом в 34 % випадкiв хворiють на рак, конкордатнiсть захворювання складає 25 % випадкiв, у двояйцевих близнюкiв - 50 %, в однояйцевих - 70 %. Лейкоз розвивається пiд впливом екзогенних та ендогенних факторiв.
В рядi випадкiв при хворобi Дауна зустрiчаються лейкемоїднi реакцiї, якi помилково дiагностуються, як лейкоз. Для карiотипу хворих з гострим лейкозом характерна поява додаткових хромосом (47 або 48), з фрагментацiєю або транслокацiєю хромосом, наявнiсть фiладельфiйської - маленької акроцентричної хромосоми (Рh) 21-ї пари, полiпоїди. При поєднаннi лейкозу з хворобою Дауна анеуплодiя непостiйна. Вважається, що 21-а хромосома має геннi локуси, що вiдповiдають за лейкопоез.
Дiагноз в типових випадках базується на даних клiнiчного обстеження. Має значення сiмейний анамнез матерi. Змiни в карiотипi пiдтверджують дiагноз захворювання.
Дiагностичнi критерiї (для новонароджених): зплощений профiль обличчя, вiдсутнiсть рефлексу Моро, м'язова гiпотонiя, косий розрiз очей, надлишок шкiри на шиї, розбовтанiсть суглобiв, диспластичний таз, диспластичнi вуха, клинодактилiя мiзинця, поперечна долонна складка(рис. 5).

Рис.5. Поперечна долонна складка.
Лiкування. В останнi роки покращилися результати лiкування дiтей iз синдромом Дауна завдяки раннiй медикаментознiй (з 2-х мiсяцiв) та психопедагогiчнiй адаптацiї дiтей. Призначають препарати, що покращують метаболiзм в мозковiй тканинi (сiднокарб, амiналон, енцефабол, ноотропiл) курсами по 2-3 мiсяцi Застосовують гормонотерапiю (тиреоїдин по 0,05-0,2 мг/добу, екстракт передньої долi гiпофiзу - префiзон, курс 30 iн'єкцiй). Масаж, гiмнастика, хiрургiчна корекцiя.Медико-генетичне консультування. Для сiм'ї де є дитина з синдромом Дауна, ризик народження хворої дитини високий, але залежить вiд цитогенетичного варiанту.
Прогноз. Тривалiсть життя в середньому 36-37 рокiв. Найбiльш частою причиною смертi є вродженi вади, здебiльшого серця.
Пренатальна дiагностика. Селективна дiагностика проводиться в групах пiдвищеного ризику: транслокацiйнi форми, коли в сiм'ї вже є хвора дитина; батьки старшi 35 рокiв, ускладнений перебiг вагiтностi.
Пiд час масової УЗД синдром Дауна можна запiдозрити на пiдставi групи ознак (форма черепа, гiпотофiя плода, вади серця, товщина шкiрної складки на шиї).
Синдроми, обумовленi частковою делецiєю аутосом
Синдроми часткової делецiї аутосом групи В. При частковiй делецiї короткого плеча 4-ї аутосоми у хворих спостерiгається маленький зрiст, "вовча паща" i "заяча губа", гiпоспадiя, гiдроцефалiя, гiпотонiя м'язiв, розумова вiдсталiсть, колобома райдужної оболонки, "крик кiшки", низьке розмiщення вух.
При синдромi "cri du chat" ("крик кiшки") виявлена делецiя короткого плеча 5-ї хромосоми. Клiнiчно даний синдром дуже полiморфний. Без своєрiдного крику дiагноз безцитогенетичного обстеження встановити не можливо. В типових випадках у дiтей кругле обличчя з гiпертелоризмом, антимонголоїднi очнi щiлини, косоокiсть, епiкант, зменшене пiдборiддя, плоский нiс, деформованi низько розмiщенi вуха, коротка шия, нижня синдактилiя, вкороченi пальцi, клинодактилiя, вродженi вади розвитку серця i статевих органiв, аномалiї нирок, розумова вiдсталiсть. Частiше хворiють дiвчатка.
Синдроми часткової делецiї 18-ї аутосоми. Часткова делецiя короткого плеча 18 аутосоми клiнiчно проявляється мiкроцефалiєю, аномалiєю очей, атрезiєю зовнiшнiх слухових каналiв, зниженням слуху, малим зростом, розумовою вiдсталiстю, м'язовою гiпотонiєю.
Для синдрому часткової делецiї довгого плеча 18-ї аутосоми характернi мiкроцефалiя, вiдставання у фiзичному i психiчному розвитку, деформацiя вушних раковин, веретеноподiбнi пальцi i т.д.
При синдромi часткової делецiї 21-ї хромосоми виявляються розумова вiдсталiсть, "антидаунiзм" (виступаюче перенiсся, великi близько розмiщенi вуха i повiки, гiпертонус м'язiв), дистрофiчнi змiни кiсток, гiпоспадiя, зменшення кiлькостi тромбоцитiв в кровi.
Лiтература1. Наследственные синдроми и медико-генетическое консультирование: Семанова Е., Демикова Н.С. и др.- Л.: с.
2. Синдром Дауна: дiагностика, опiка, запобiгання /Пiд ред. Л.С. Евтушок.- Рiвне, 2003.- 165 с.
3. Смiян I.С., Банадига Н.В., Багiрян I.О. Медична генетика дитячого вiку. -Тернопiль:Укрмедкнига, 2003. - 186 с.
Контрольнi запитання
1. Що вiдносять до хромосомних захворювань?
2. Назвiть на якi групи роздiляються дiагностичнi ознаки хромосомних аномалiй?
3. Етiопатогенез синдрому Клайнфельтера.
4. Клiнiка синдрому Клайнфельтера.
5. Дiагностичнi критерiї синдрому Клайнфельтера.
6. Лiкування синдрому Клайнфельтера.
7. Етiопатогенез синдрому Шерешевського-Тернера.
8. Клiнiка синдрому Шерешевського-Тернера.
9. Дiагностичнi критерiї синдрому Шерешевського-Тернера.
10. Лiкування синдрому Шерешевського-Тернера.
11. Етiологiя, клiнiка, лiкування синдрому Патау.
12. Етiологiя, клiнiка, лiкування синдрому Едварда.
13. Етiологiя i патогенез хвороби Дауна.